The unwritten rules of mapping data include avoiding too much data and clutter, and not using too much colour. This blog often violates those rules, and when it comes to visualising journeys to work, I think we can learn a lot about cities with somewhat cluttered colourful animated maps.
This post maps journeys to work in Melbourne, using data from the 2016 census. I will look at which types of home-work pairs have different public, private and active transport mode shares and volumes.
Although this post will focus on Melbourne, I will include a brief comparison to Sydney at the end.
Where are public transport journeys to work in Melbourne?
First I need to explain the maps you are about to see.
So that I can show mode shares, I’ve grouped journeys between SA2s (which are roughly the size of a suburb). Lines are drawn from the population centroid of the home SA2 (thin end) to the employment centroid of the work SA2 (thicker end). Centroids are calculated as the weighted average location residents/jobs in each SA2 (using mesh block / destination zone data). This generally works okay for urban areas, but be aware that actual trips will be distributed across SA2s, and some SA2s on the urban fringe are quite large.
The thickness of each line at the work end is roughly proportional to the number of journeys by the mode of interest between the home-work pair (refer legends), but it’s difficult to use a scale that is meaningful for smaller volumes. Unfortunately there’s only so much you can do on a 2-D chart.
I’ve not drawn lines where there are fewer than 50 journeys in total (all modes), or where there were no journeys of the mode that is the subject of the map. This threshold of 50 isn’t perfect either as SA2s are not consistently sized within and between cities, so larger SA2s are more likely to generate lines on the map.
To try to help deal with the clutter, I’ve made the lines somewhat transparent, and also animated the map to highlight trips with different mode share intervals. For frames showing all lines, the lines with highest mode share are drawn on top.
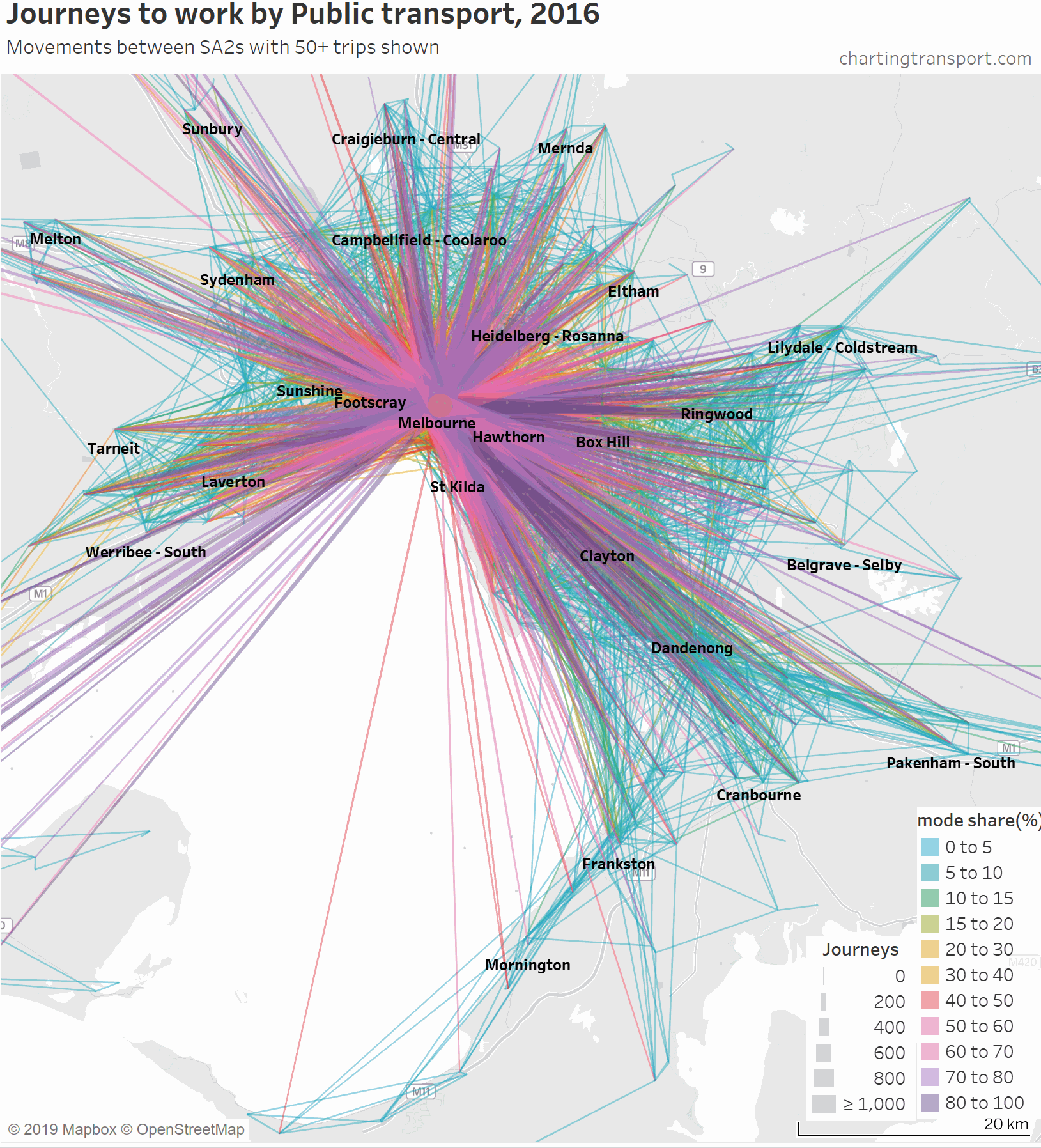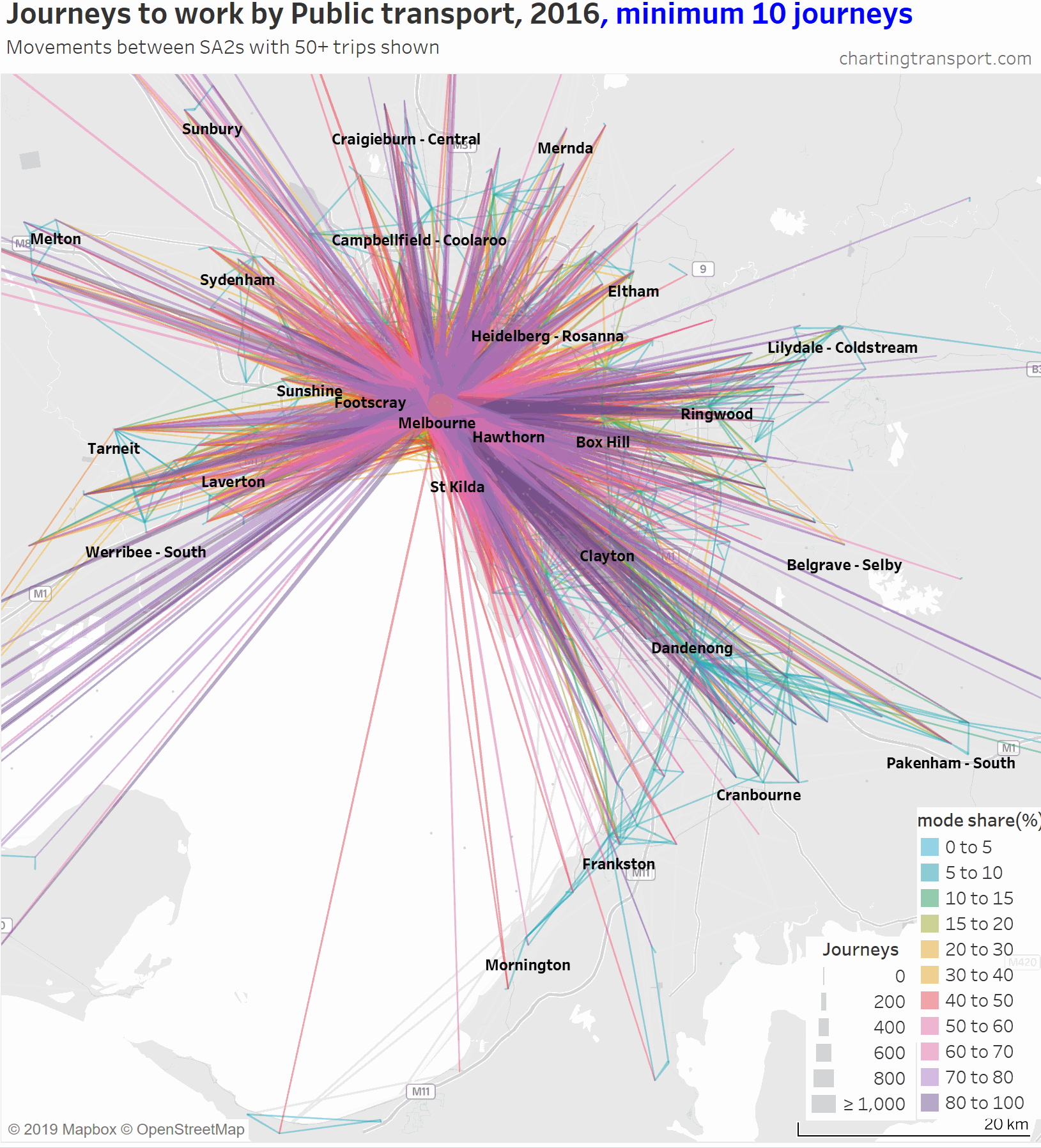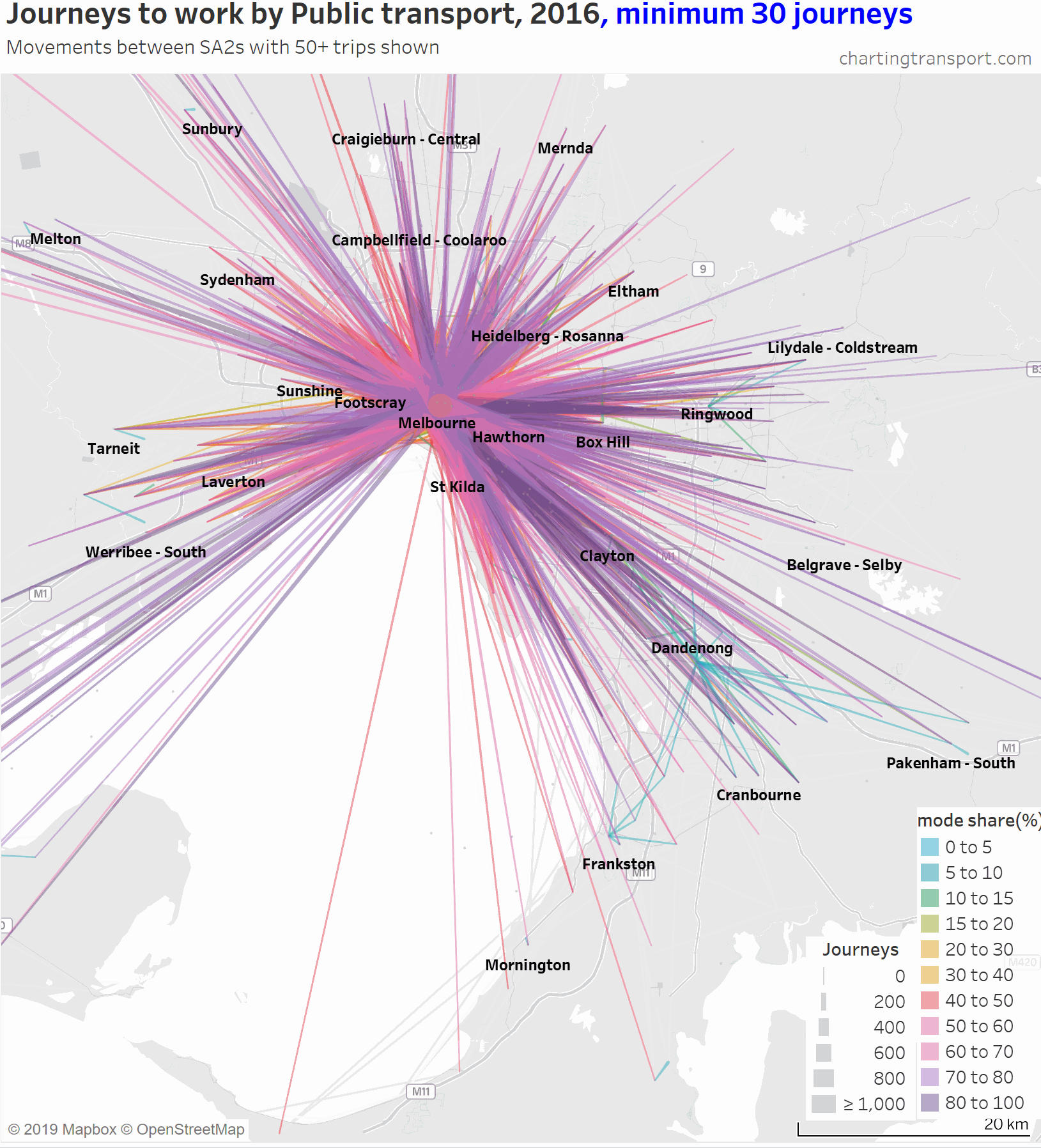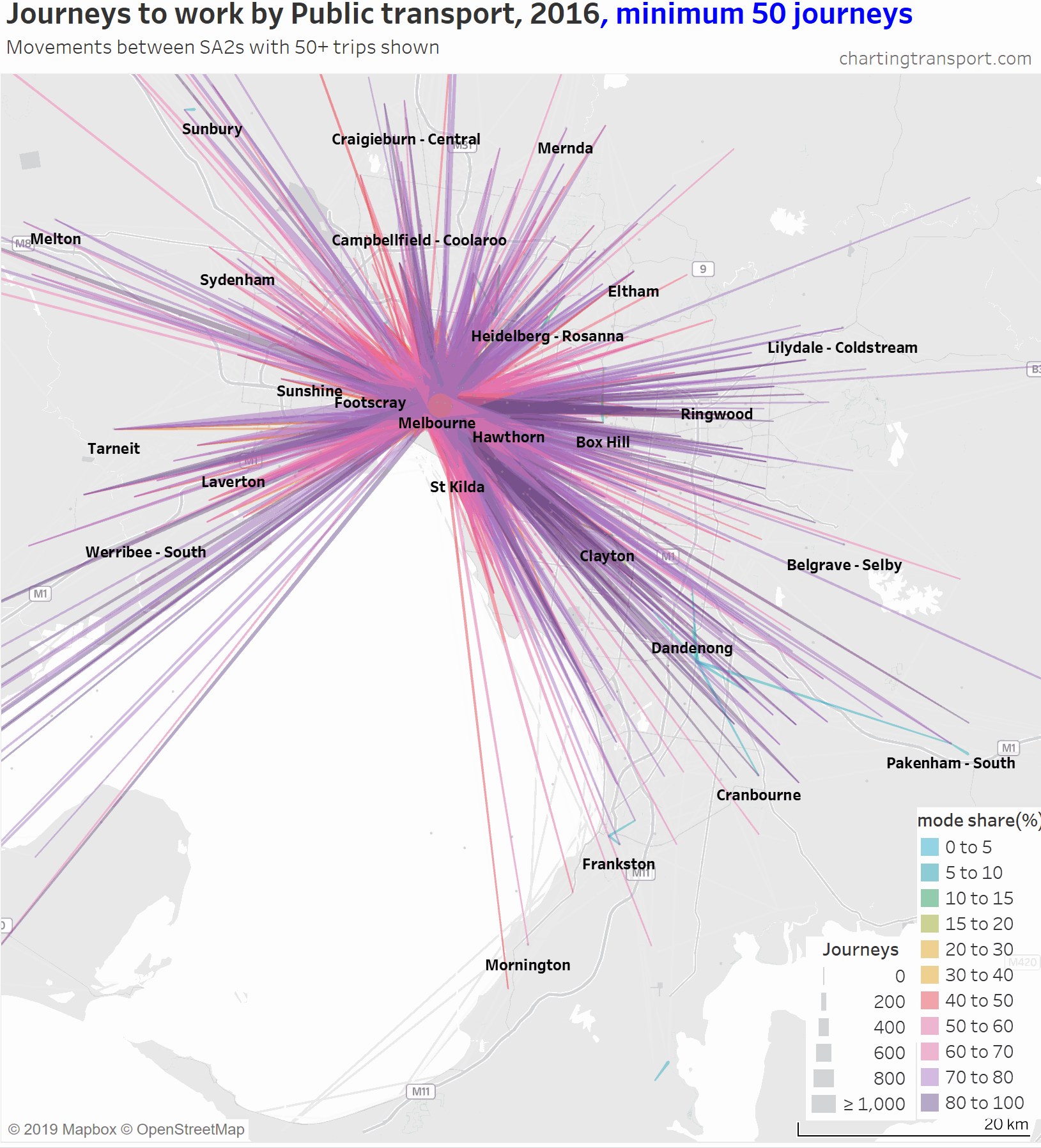
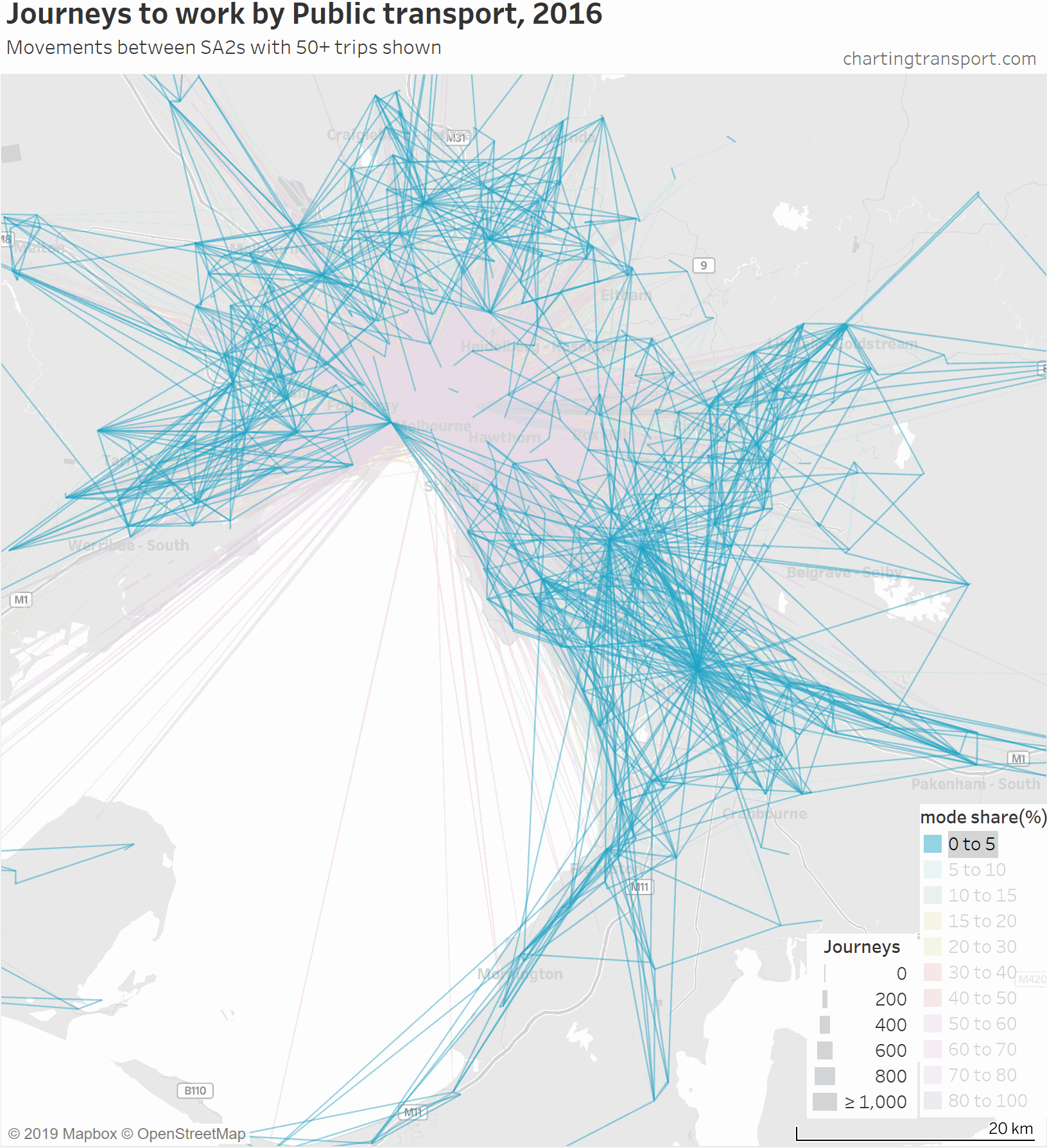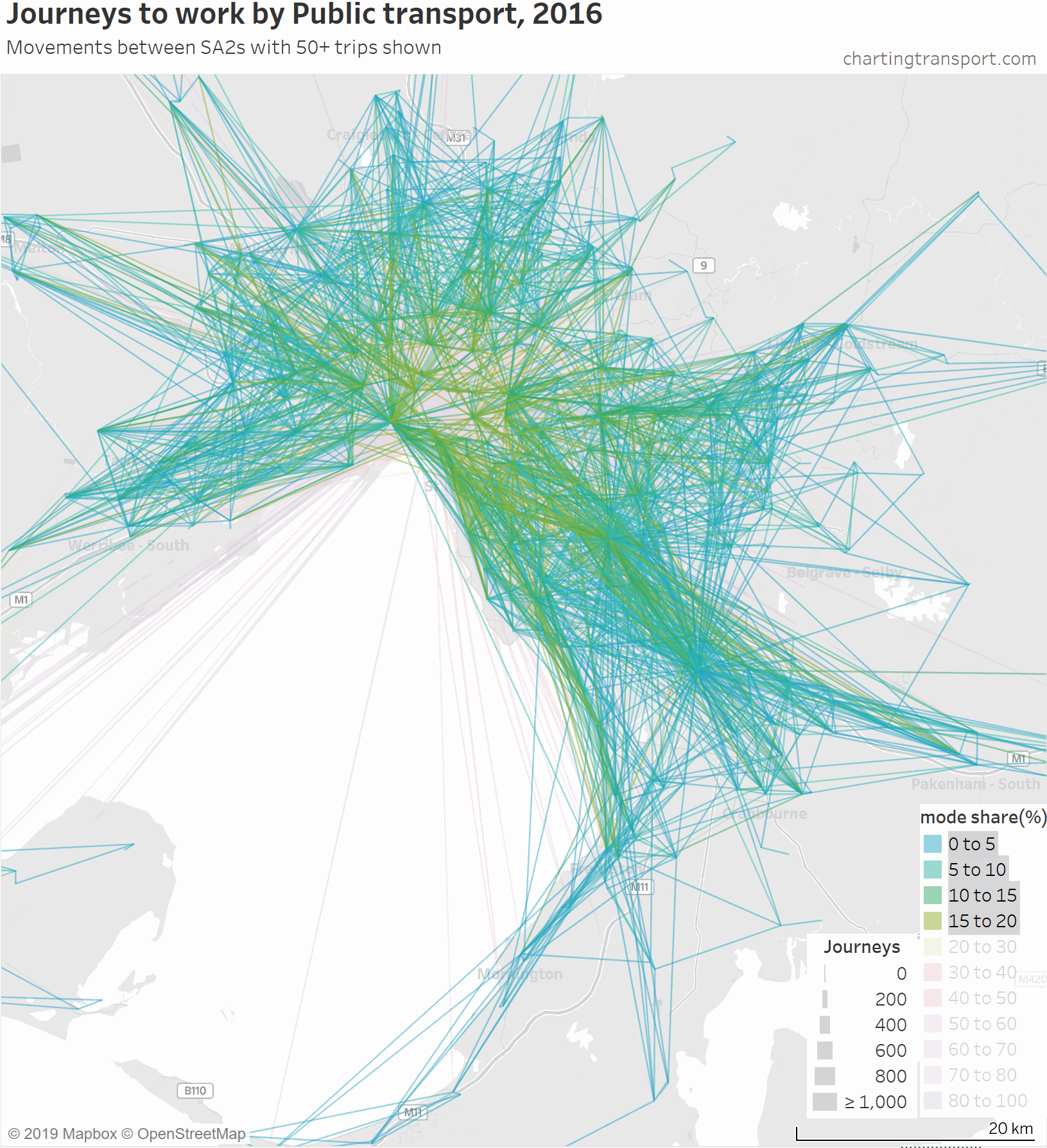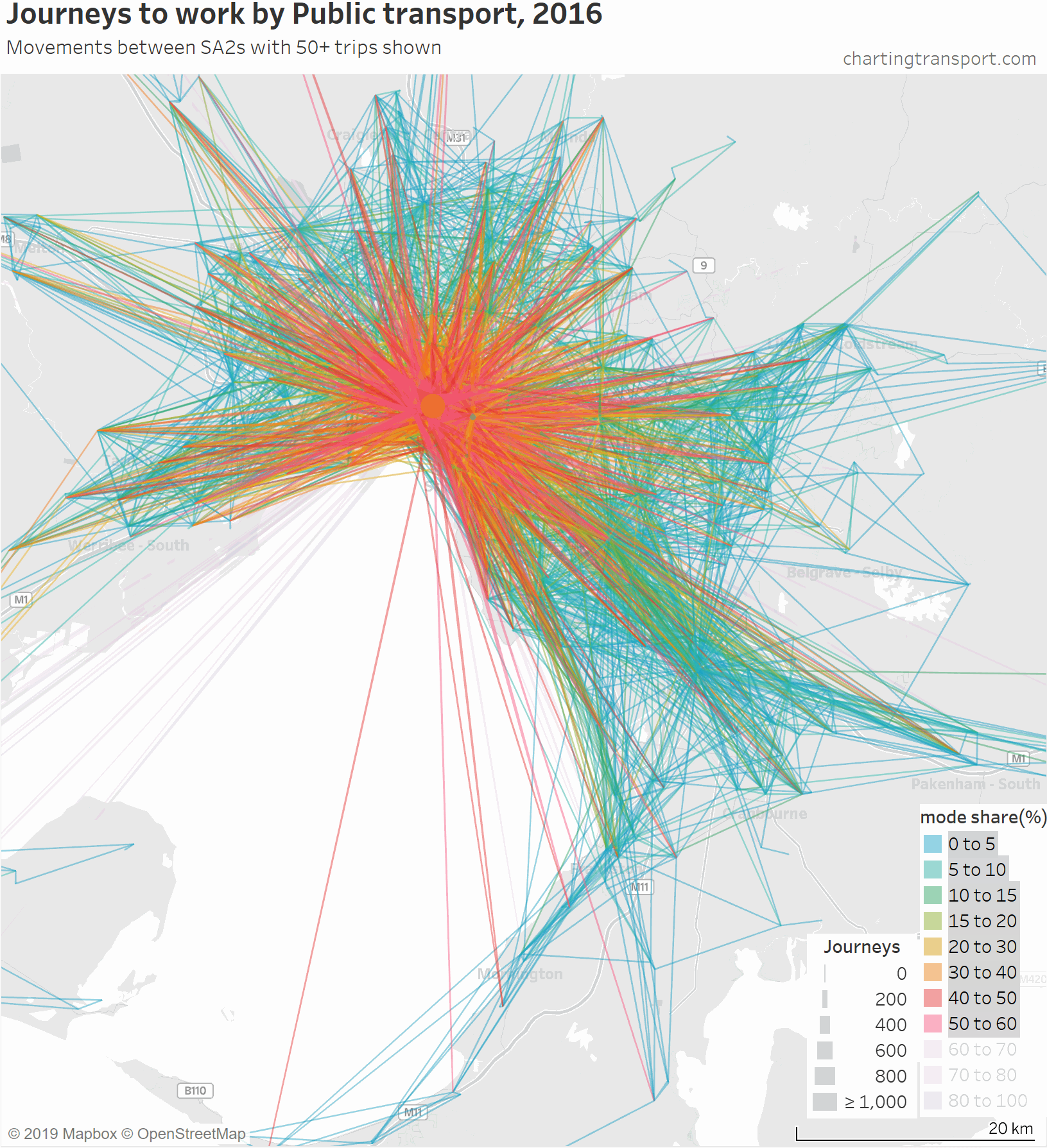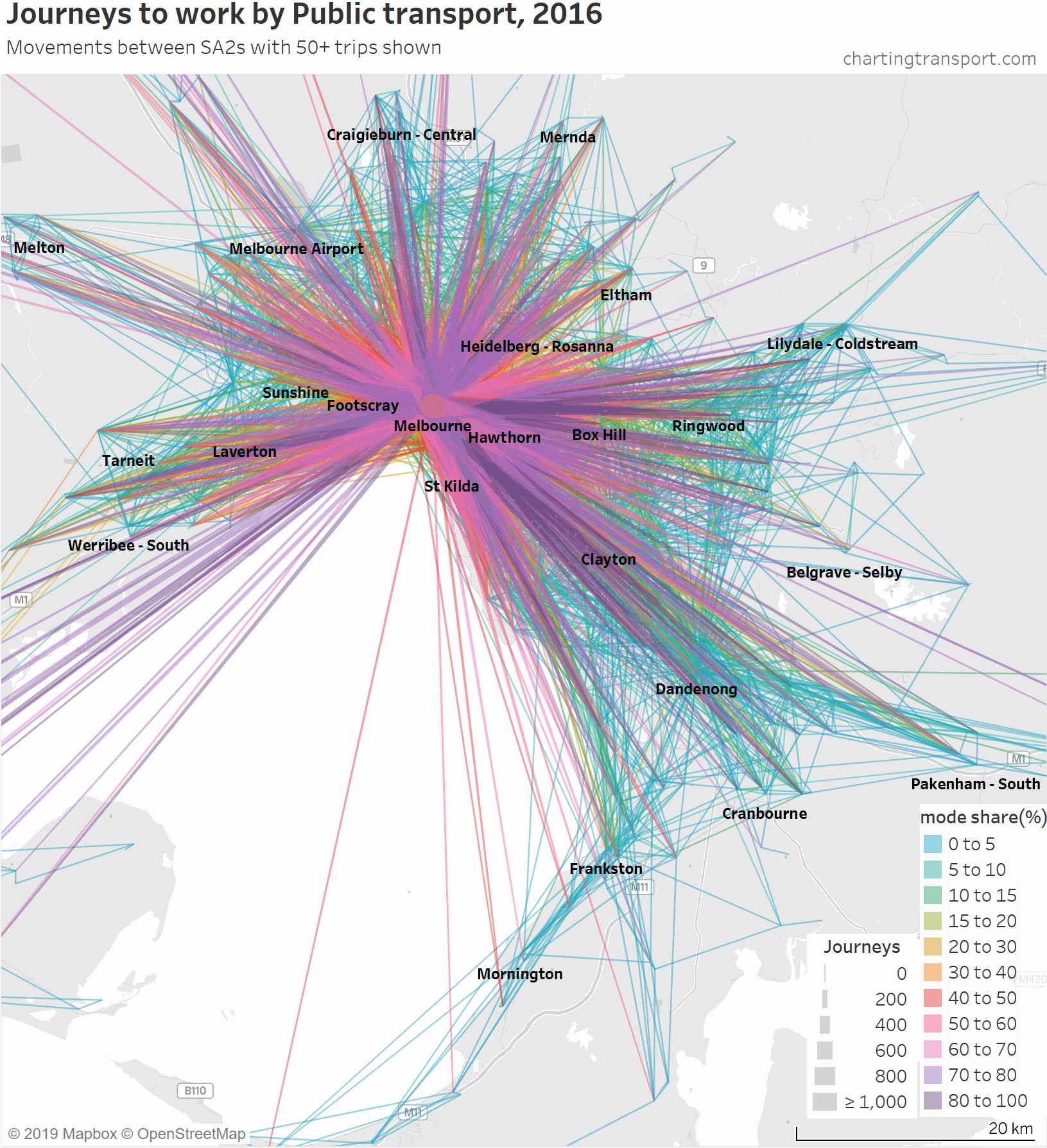
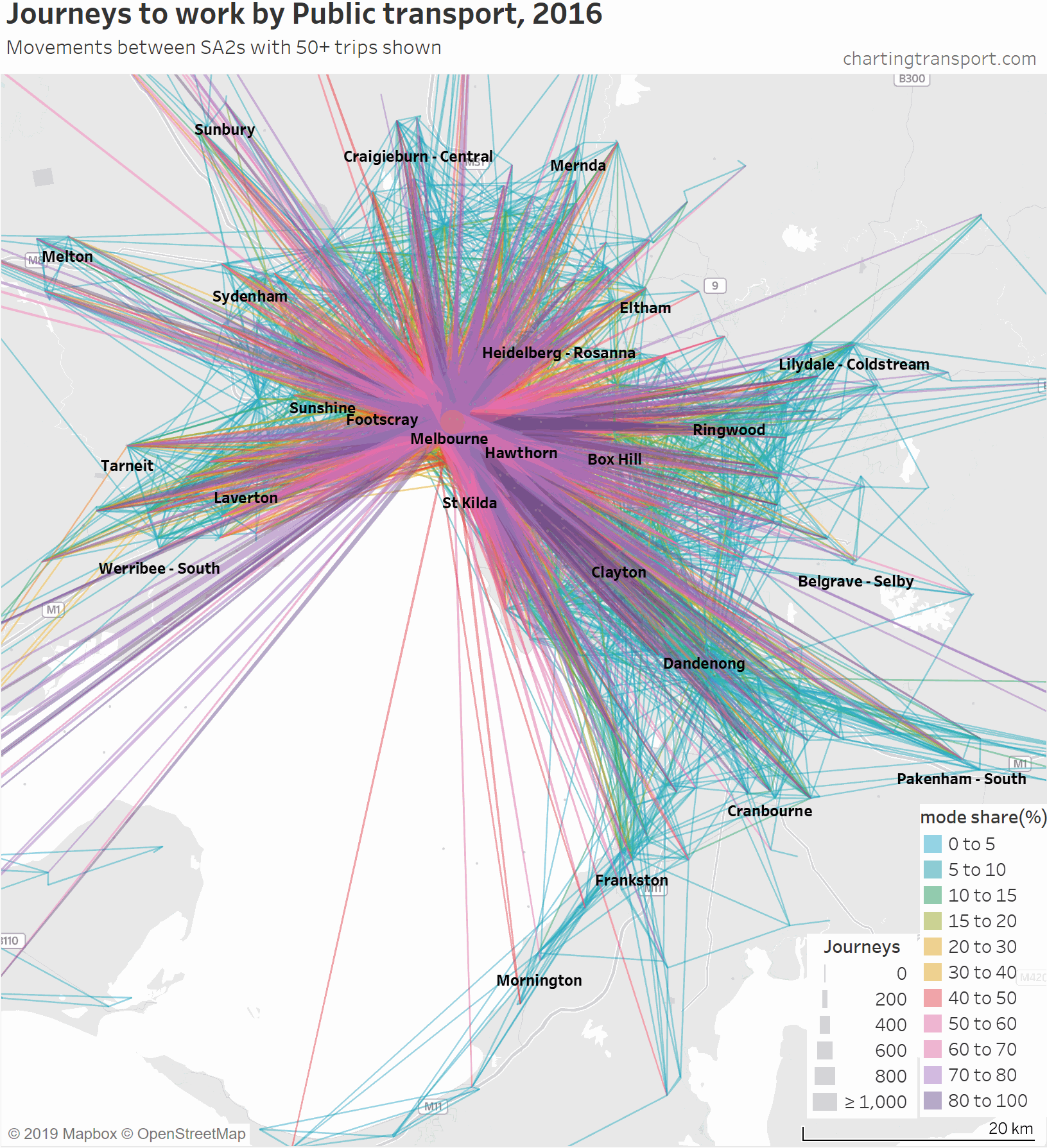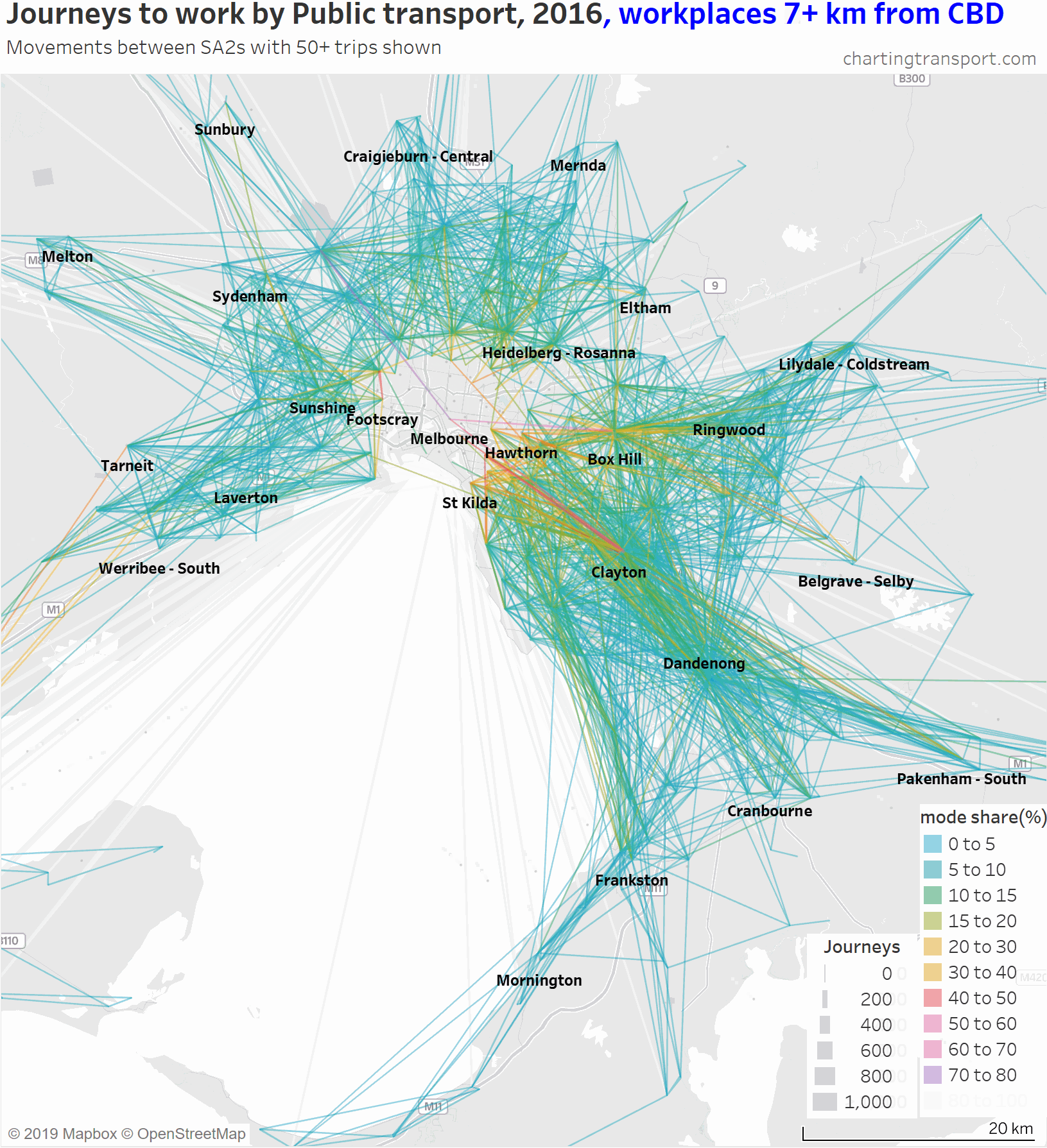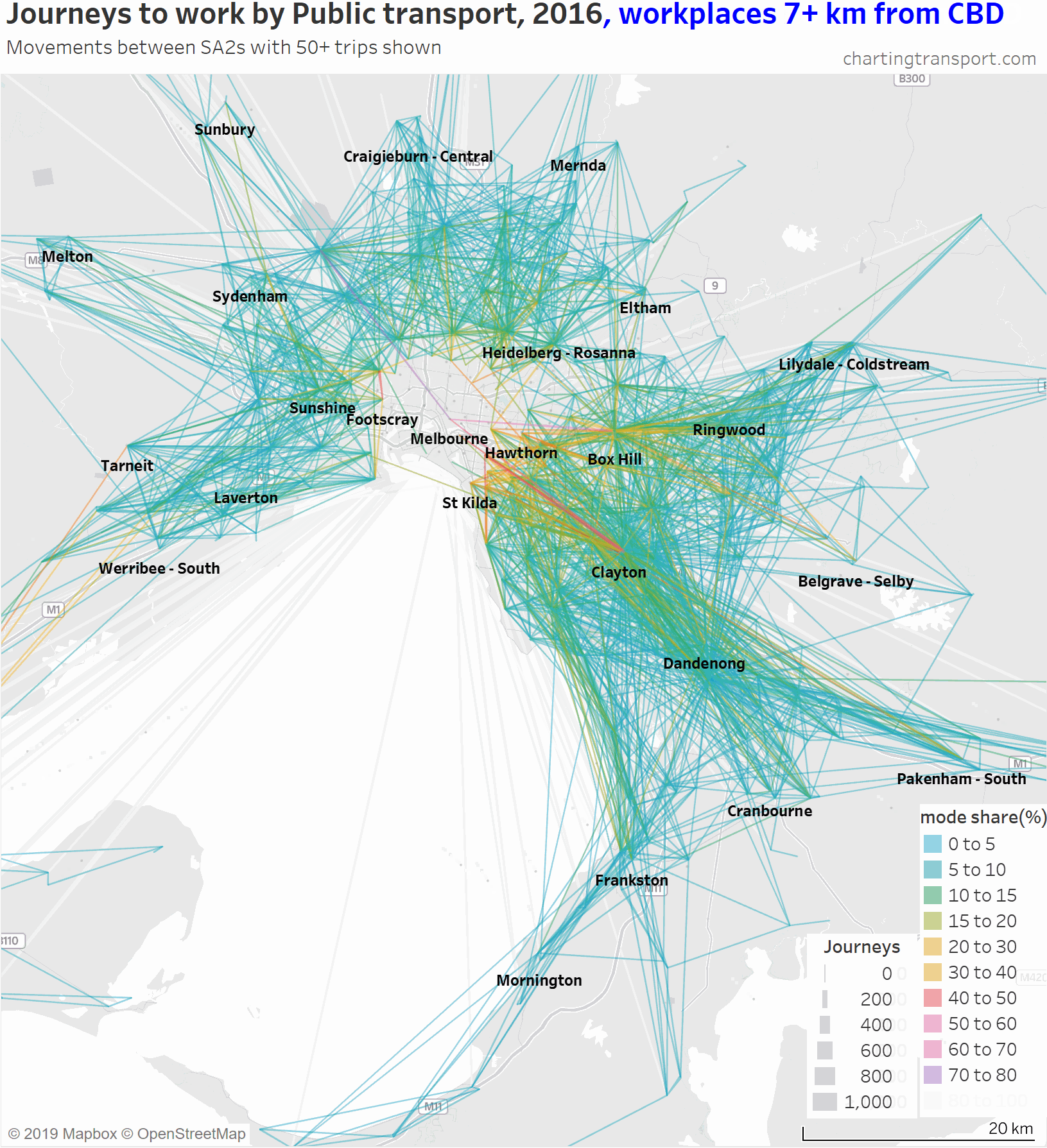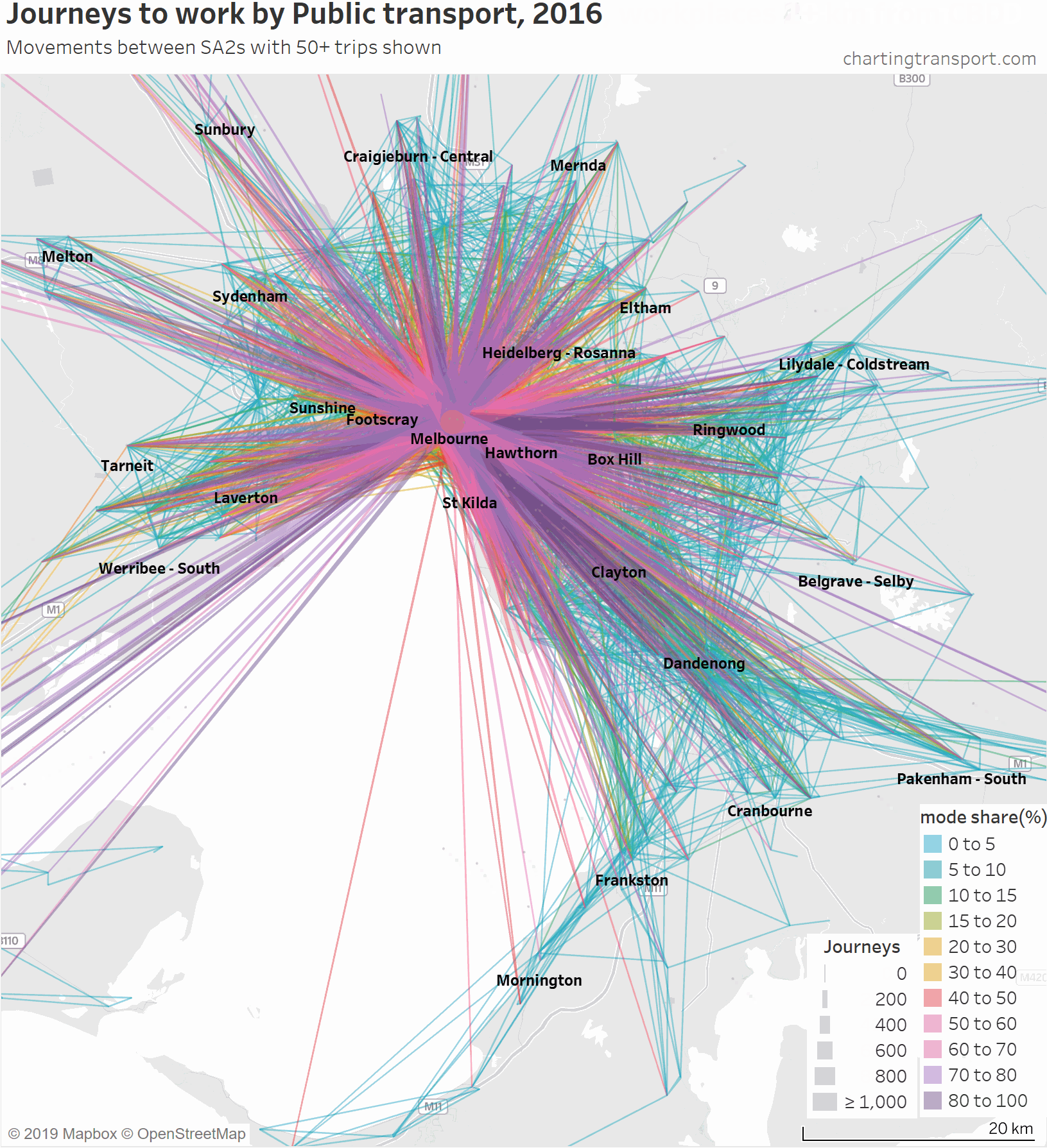
So here is an animated map showing public transport journeys to work in Melbourne, by different mode share ranges and overall:
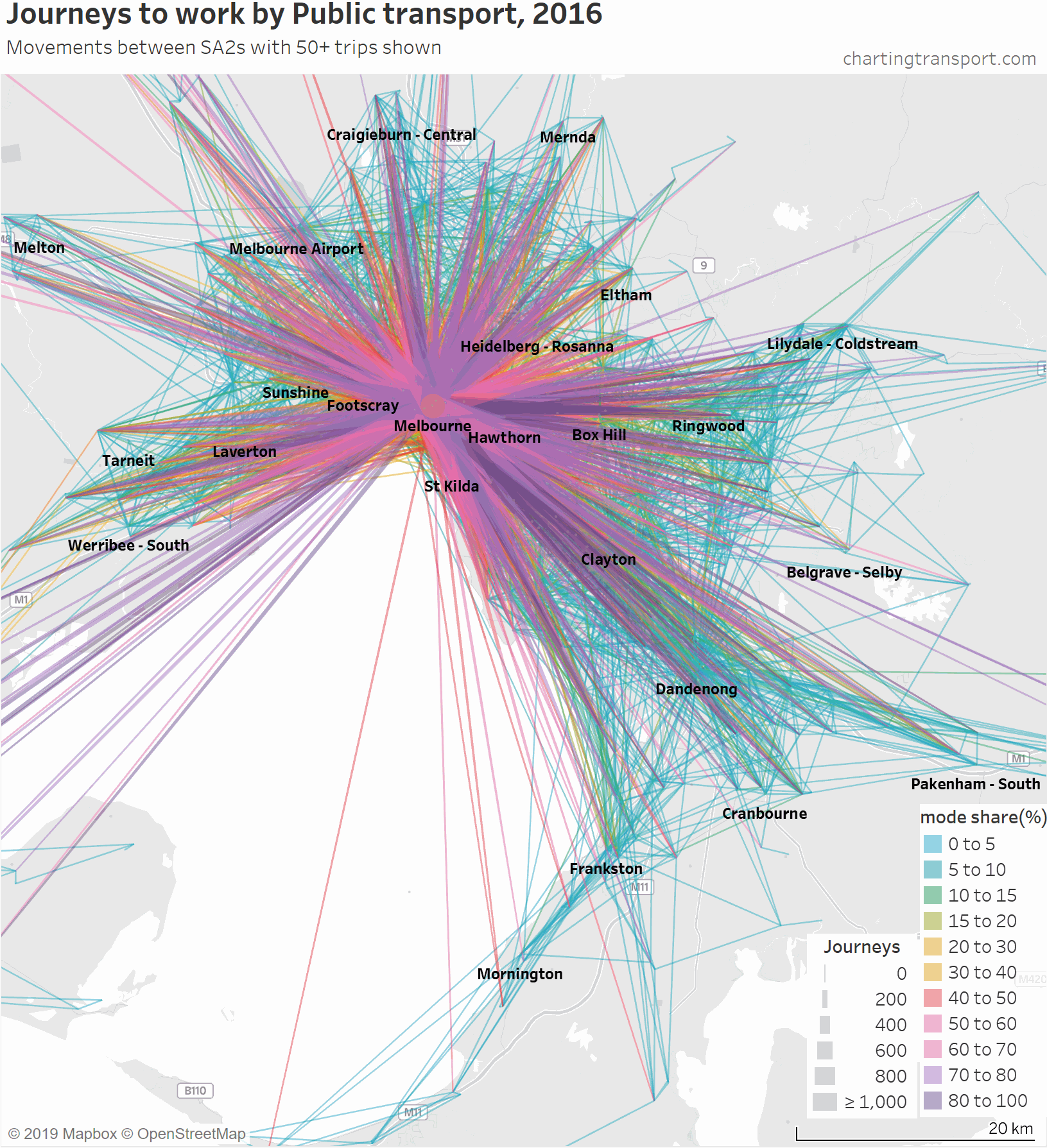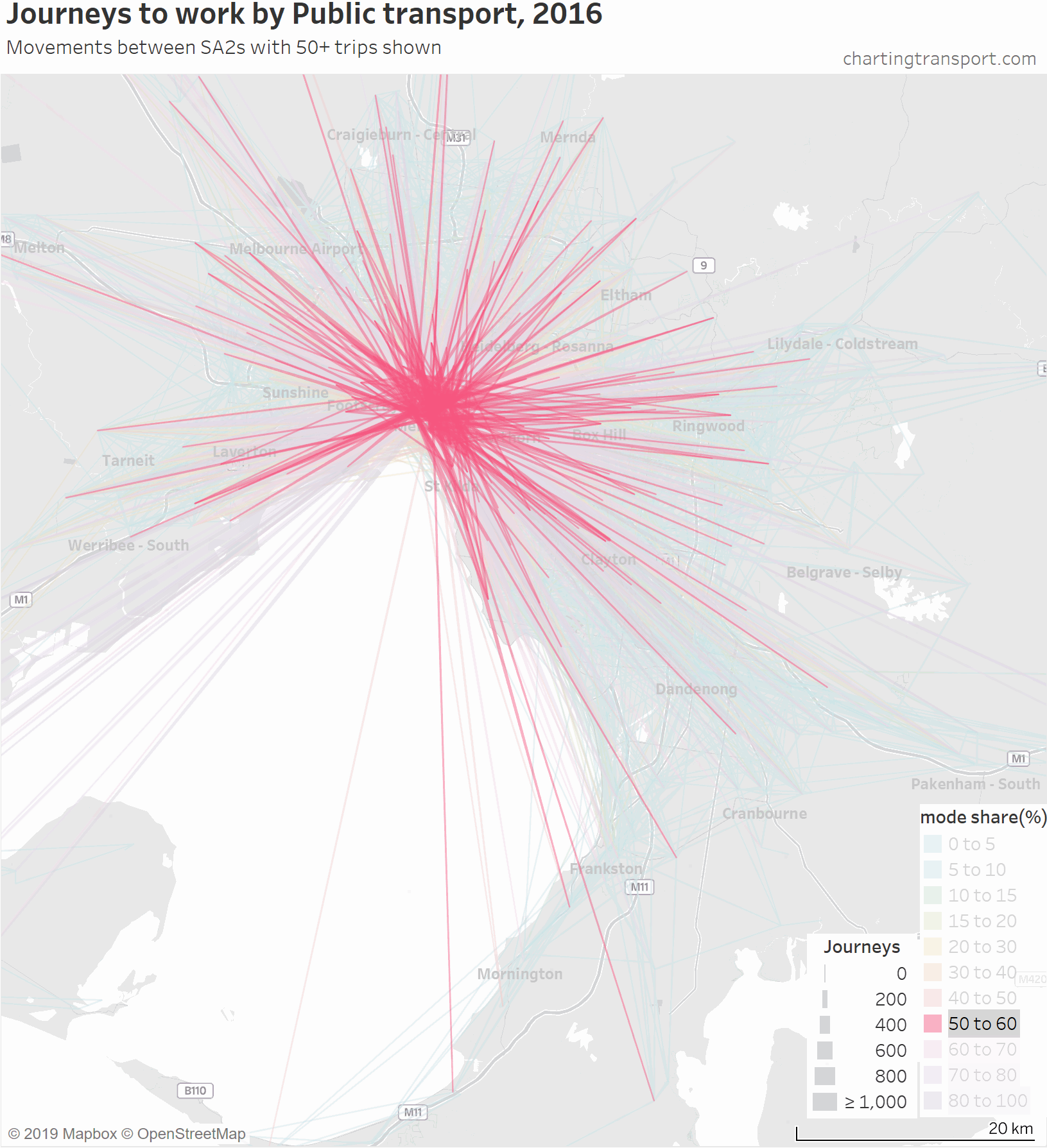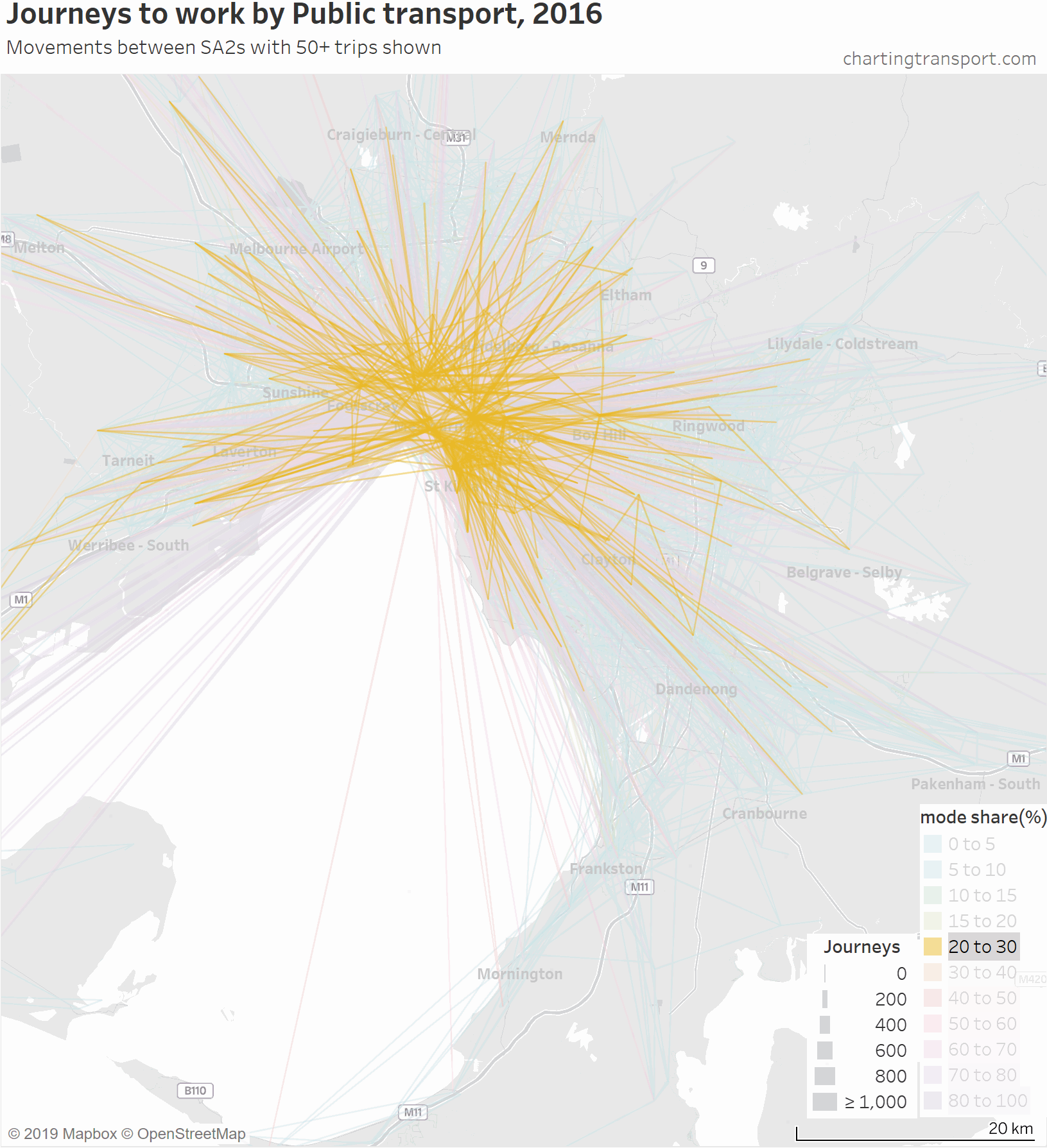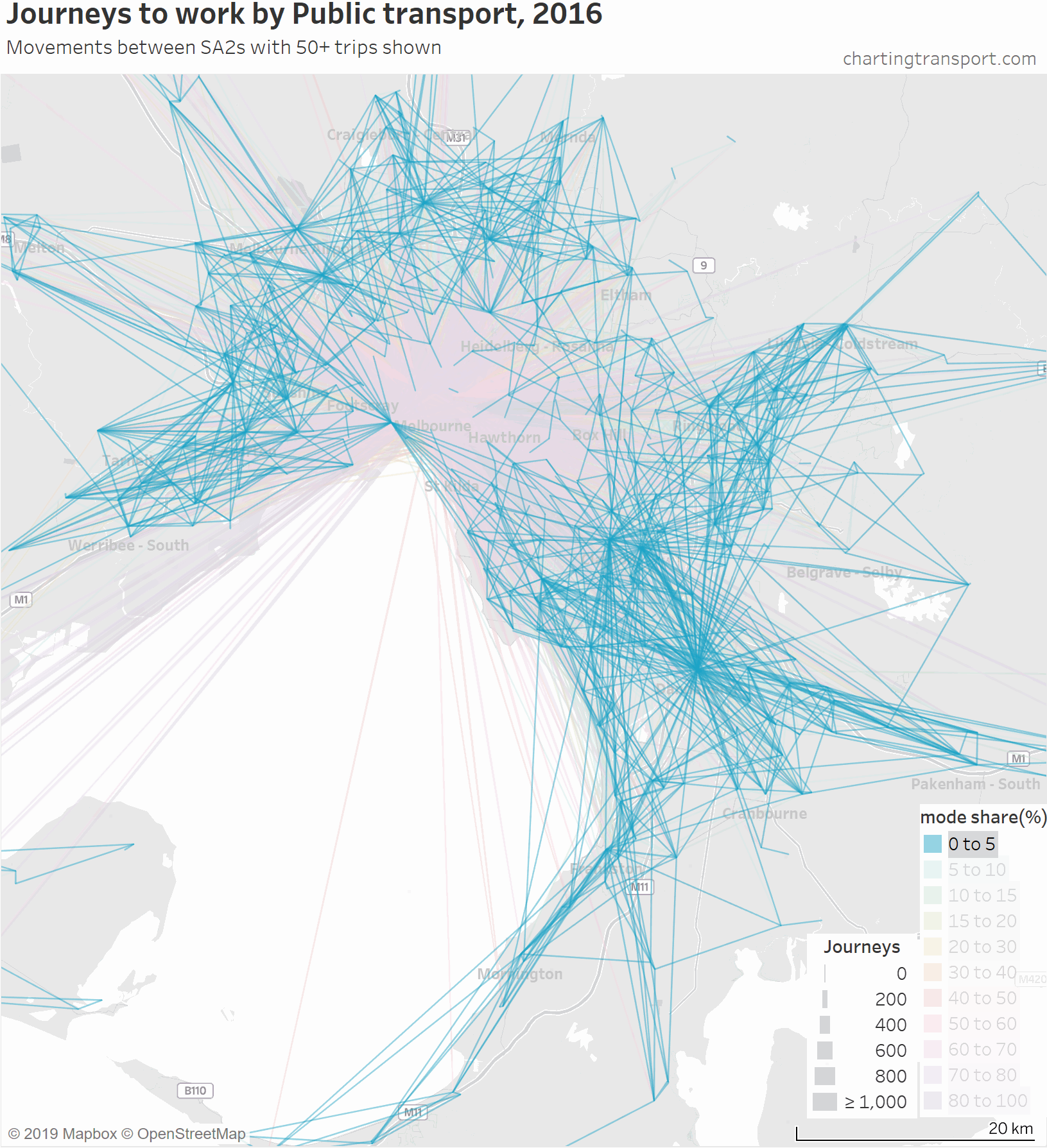
Technical note: I have included journeys to work that are internal to an SA2. Usually these appear as simple circles, but sometimes they appear as small teardrops where the population and employment centroids are sufficiently far apart.
You can see that the highest PT shares and largest PT volumes are for journeys to the central city, and generally from SA2s connected to Melbourne CBD by train (including many outer suburbs).
As the animation moves to highlight lower PT mode share ranges, the lines become a little less radial, a little shorter on average, and the lowest (non-zero) PT mode shares are mostly for suburban trips.
A notable exception is journeys to Port Melbourne Industrial SA2 (also known as Fishermans Bend), which is located at the junction of two major motorways and is remote from rapid public transport (it does however have a couple of high frequency bus lines from the CBD).
The lowest PT mode shares are seen for trips around the outer suburbs. The maps above unfortunately aren’t very good at differentiating small volumes. The following animated map shows public transport journeys with a filter progressively applied to remove lines with small numbers of public transport journeys (refer blue text in title):
You can see that most of the outer suburban lines quickly disappear as they have very small volumes. Inter-suburban lines with more than 50 public transport journeys go to centres including Dandenong, Clayton, Box Hill, and Heidelberg.
Here’s another animation that builds up the map starting with low public transport mode share lines, and then progressively adds lines with higher public transport mode shares:
As an aside, here is a chart showing journeys to work by straight line distance (between SA2 centroids), public transport mode share, work distance from the CBD and home-work volume:
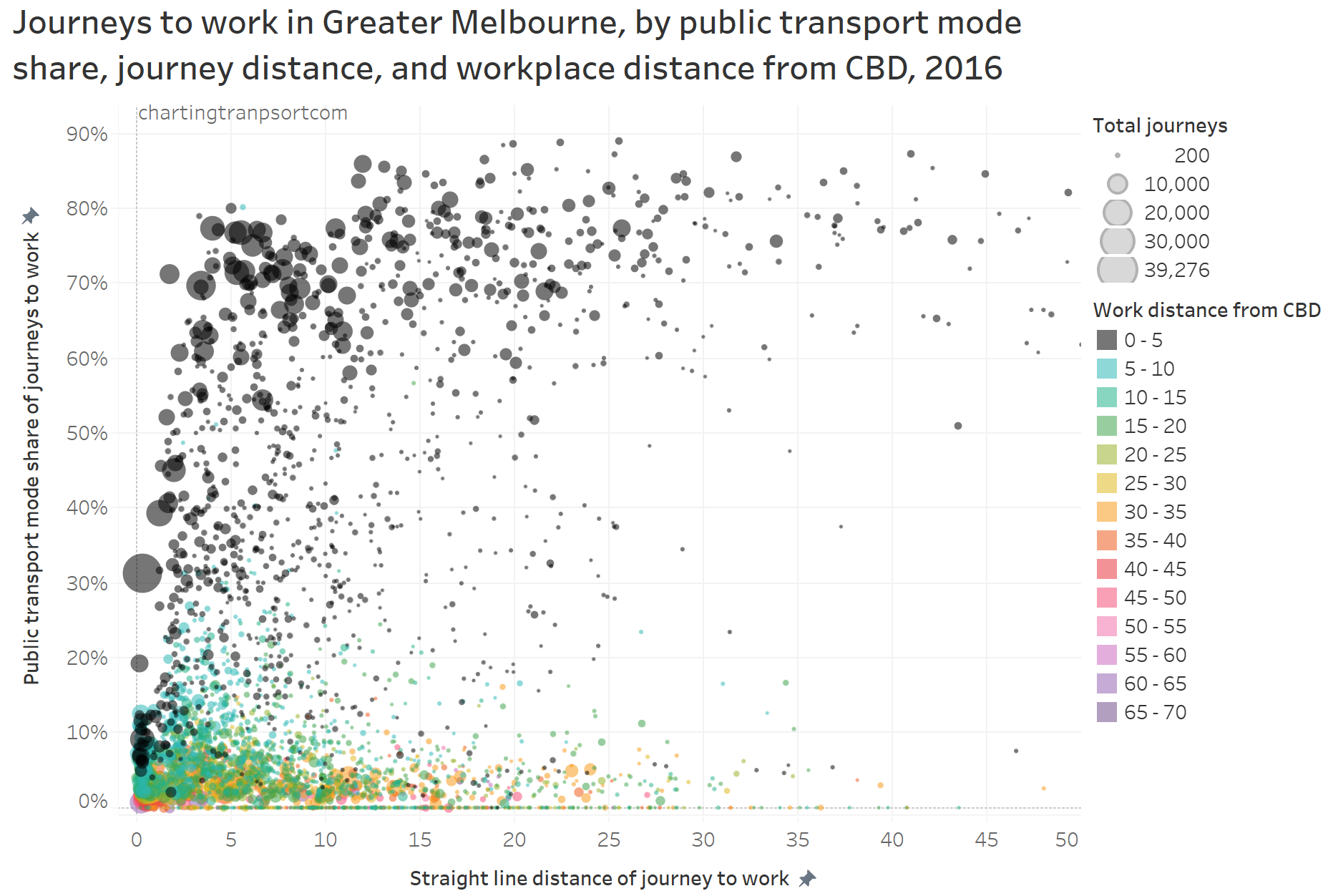
The black dots represent journeys to the inner 5km of the city, and you can see public transport has a high mode share of longer trips. Public transport mode share falls away for shorter journeys to the inner city as people are more likely to use active transport. A dot on the top left of the curve is 8,874 journeys from Docklands to Melbourne – which benefits from the free tram zone and the distances can be 1-2 km. Most of the longer journeys with low public transport mode share are to workplaces remote from the CBD (coloured dots).
Another way to deal with the clutter of overlapping lines around the CBD is to progressively remove lines to workplaces in and around the CBD. Here is another animated map that does exactly so that you can better see journeys in the nearby inner and middle suburbs.
As you strip away the CBD and inner suburbs, you lose most lines with high public transport mode shares and volumes. However some high public transport mode share lines remain, including the following outbound journeys:
- Melbourne (CBD) to Melbourne Airport: 72% of 64 journeys
- Melbourne (CBD) to Box Hill: 66% of 76 journeys
- Melbourne (CBD) to Clayton: 57% of 82 journeys
- South Yarra – East to Clayton: 57% of 173 journeys
Just keep in mind that these are all very small volumes compared to total journeys in Melbourne.
You might have noticed on the western edge of the map some yellow and orange lines from the Wyndham area (south-west Melbourne) that go off the map towards the south west. These journeys go to Geelong.
Here’s a map showing journeys around Geelong and between Geelong and Greater Melbourne (journeys entirely within Greater Melbourne excluded):
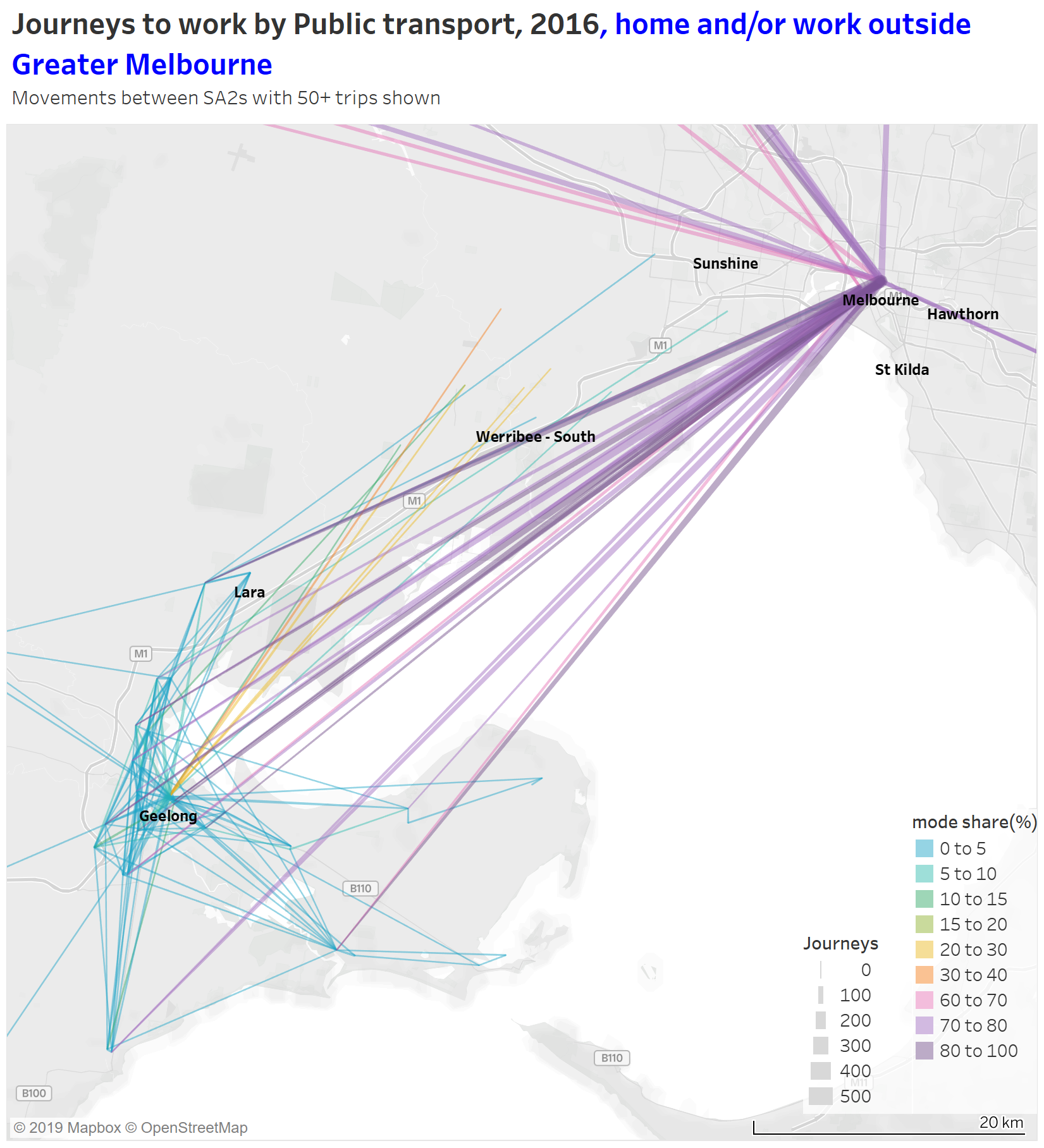
You can see very high public transport mode shares for journeys from the Geelong and Bellarine region to the Melbourne CBD and Docklands (and fairly large volumes), but no lines to Southbank, East Melbourne, Parkville or Carlton – all more remote from Southern Cross Station, the city terminus for regional trains.
(The other purple lines to the CBD are from Ballarat, Bacchus Marsh, Daylesford, Woodend, Kyneton, Castlemaine, Kilmore-Broadford and Warragul, with at least 60 journeys each.)
You can also see those orange and yellow lines from the Wyndham area to central Geelong, being mode shares of 20-40%. The Geelong train line provides frequent services between Tarneit, Wyndham Vale, and Geelong, and has proved reasonably popular with commuters to Geelong (frequency was significantly upgraded in June 2015 with the opening of the Regional Rail Link, just 14 months before the census of August 2016).
However, public transport mode shares for travel within Greater Geelong are very small – even for SA2 that are connected by trains. This might reflect Geelong train station being on the edge of its CBD, relatively cheap parking in central Geelong, limited stopping frequency at some stations (many at 40 minute base pattern), and/or limited walk-up population catchments at several of Geelong’s suburban train stations.
Does public transport have significant mode share for cross-suburban journeys to work?
To search for cross-suburban journeys with relatively high public transport mode shares, here is a map that only shows lines with public transport mode shares above 20% between homes and workplaces both more than 5 km from the CBD (yes, those are arbitrary thresholds):
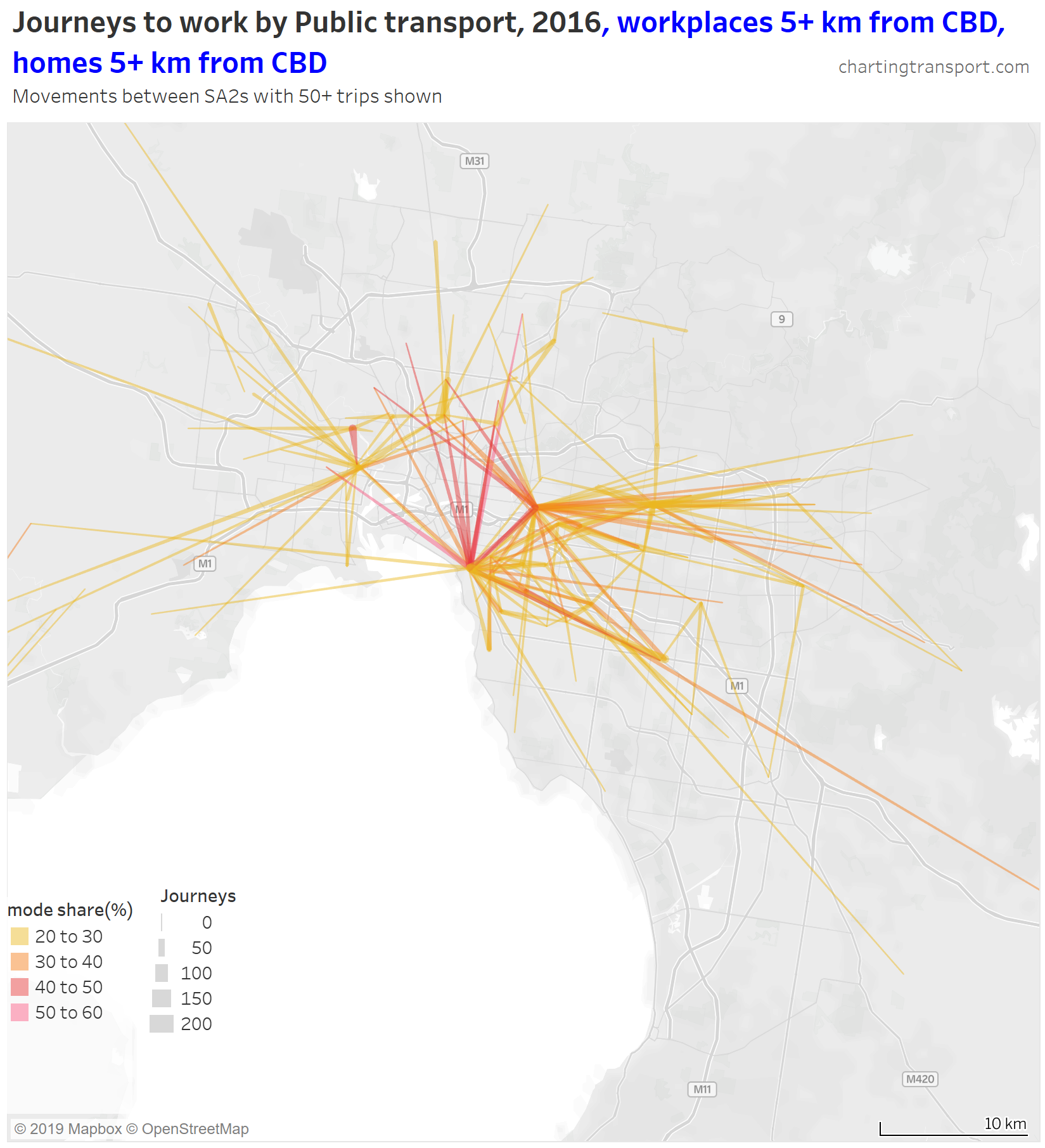
Of these journeys, the highest mode shares are for journeys from the inner northern suburbs to St Kilda and Hawthorn. There’s also a 49% mode share from Footscray to Maribyrnong (connected by frequent trams and buses).
The tear drop to the north of the city is 114 people who used PT from Coburg to Brunswick (connected by two tram routes and one train route).
Most of the other links on this map are fairly well aligned with train, tram, or SmartBus routes, suggesting high quality services are required to attract significant mode shares.
But these trips are a tiny fraction of journeys to work around Melbourne. In fact 3.0% of journeys to work in Melbourne were by public transport to workplaces more than 5 km from the CBD. The same statistic for Sydney is more than double this, at 7.3%.
What about private transport journeys?
Firstly, here’s a map showing private transport mode shares and volumes, building up the map starting with low private mode share lines.
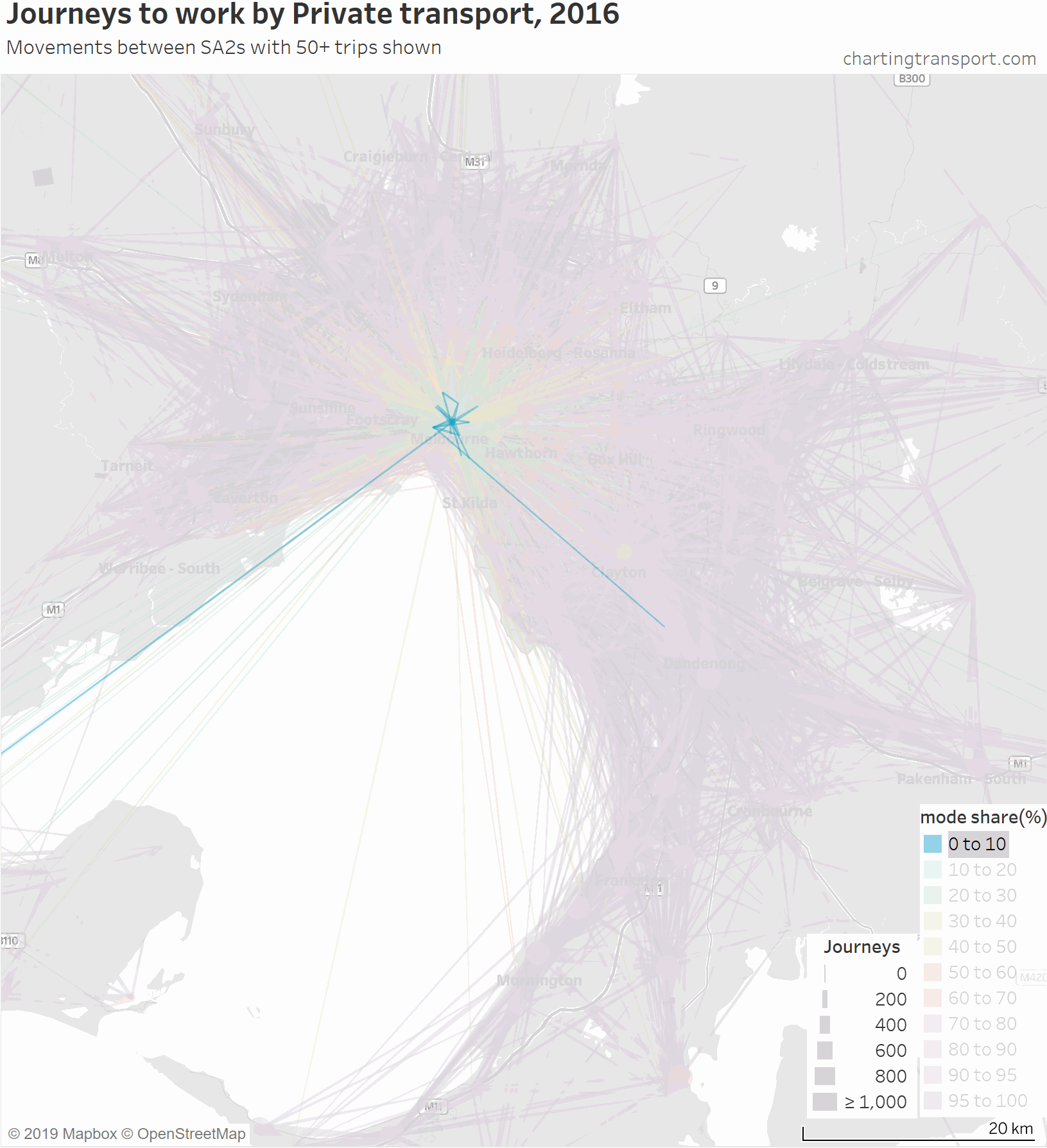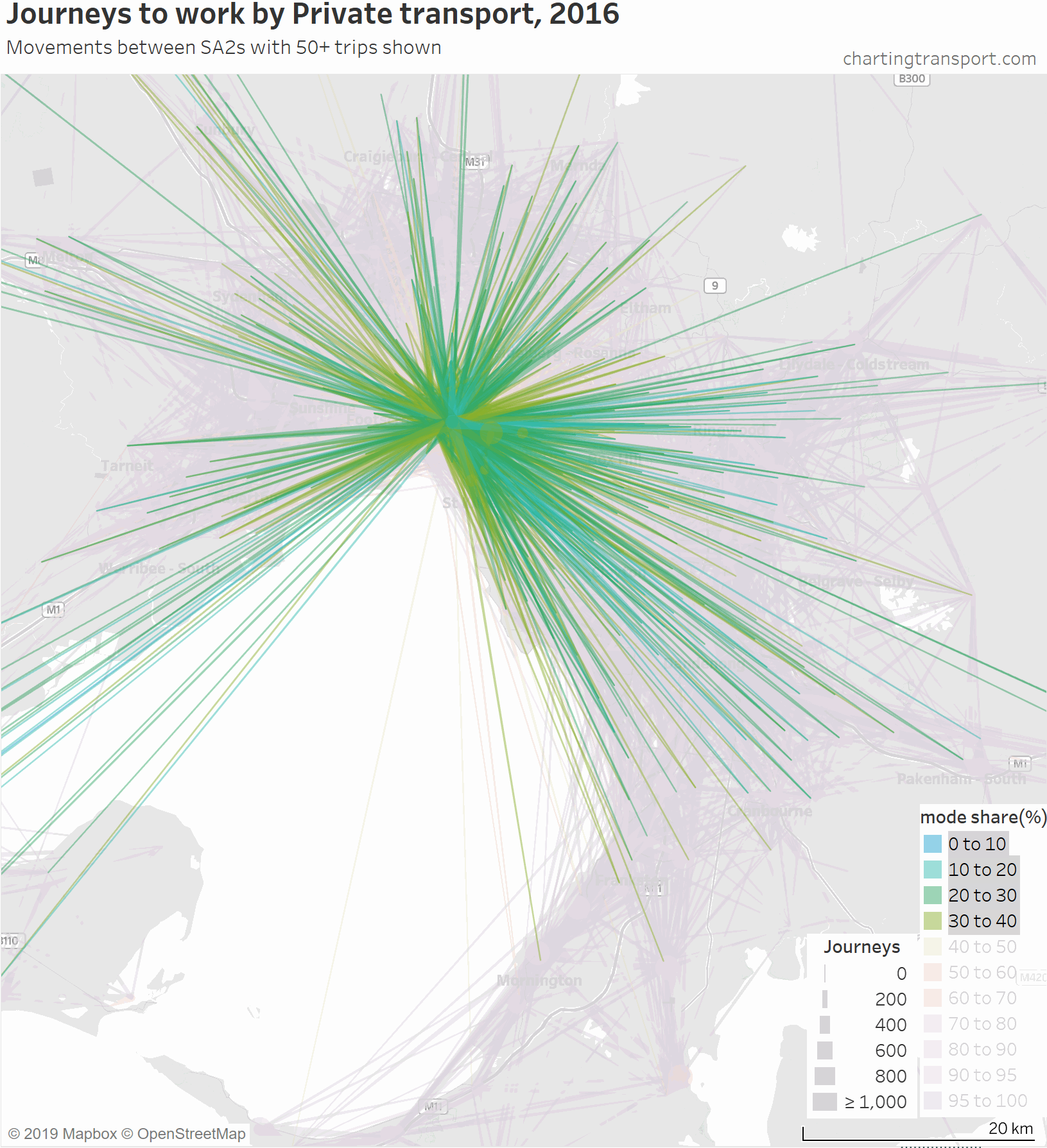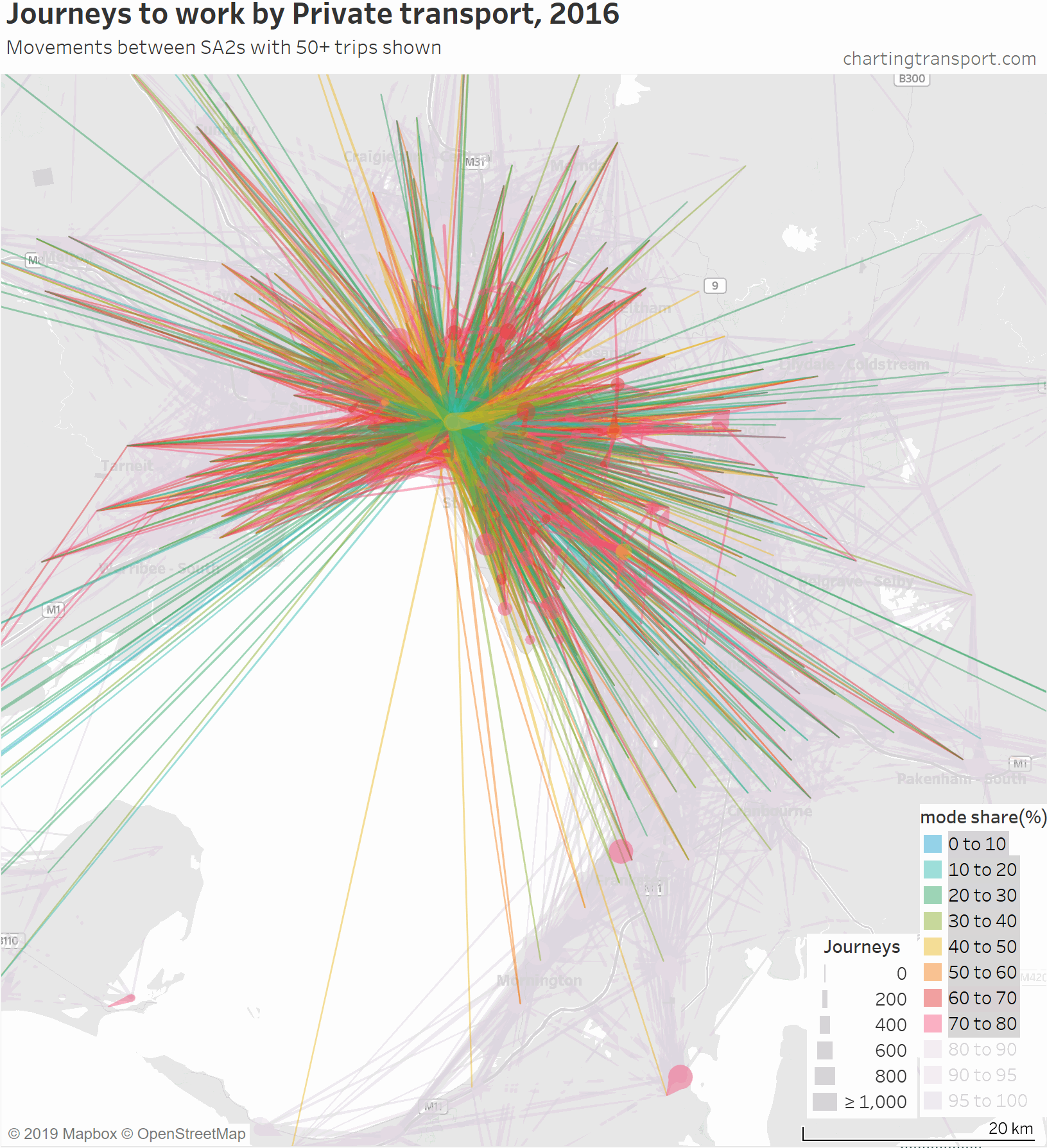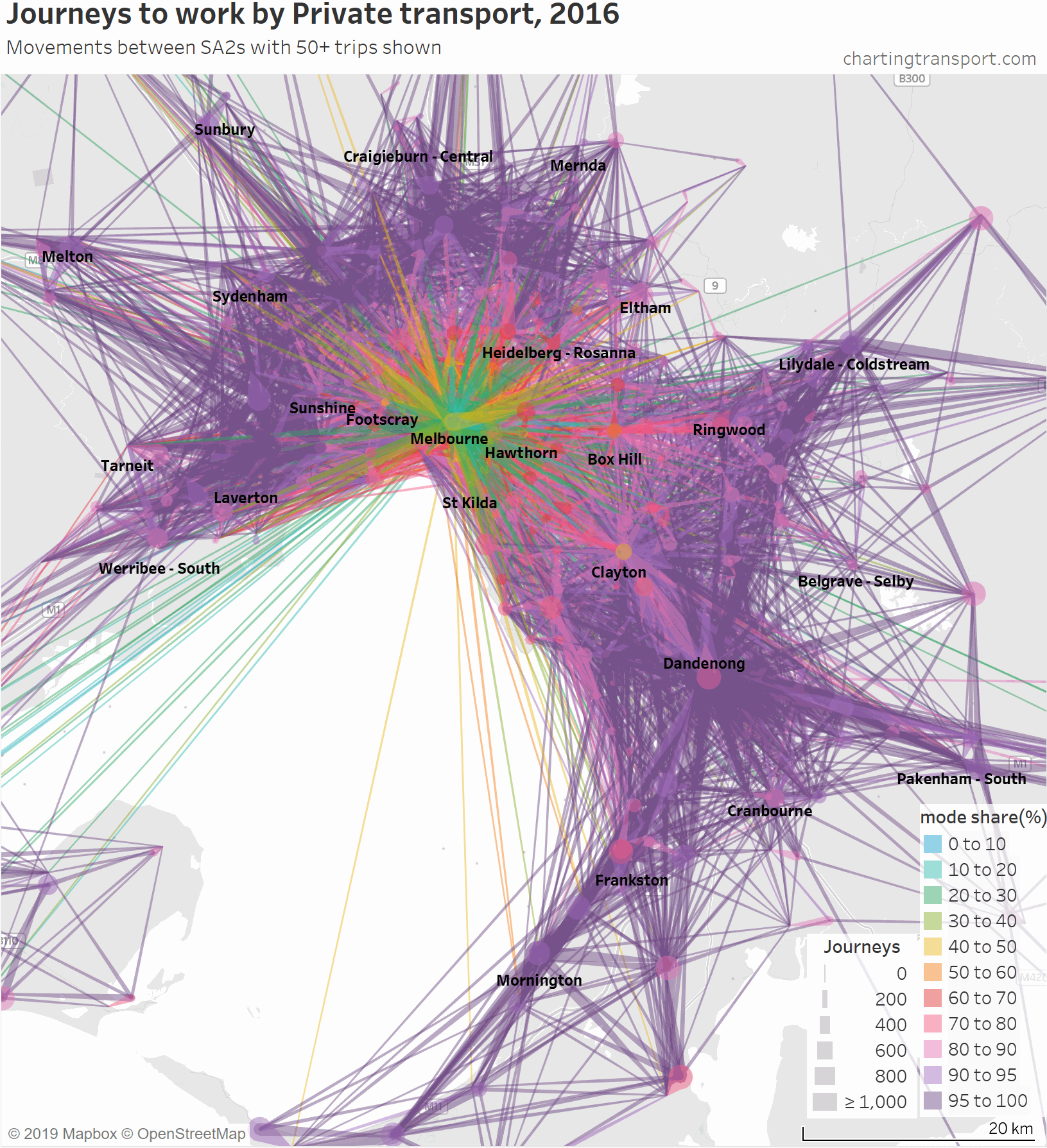
The links with lowest private transport mode shares are very radial as you might expect (pretty much the inverse of the public transport maps). Progressively less radial lines get added to the map before there is a big bang when the final private transport mode share band of 95-100% gets added, with large volumes of outer suburban trips.
For completeness, here’s an animation that highlights each mode share range individually.
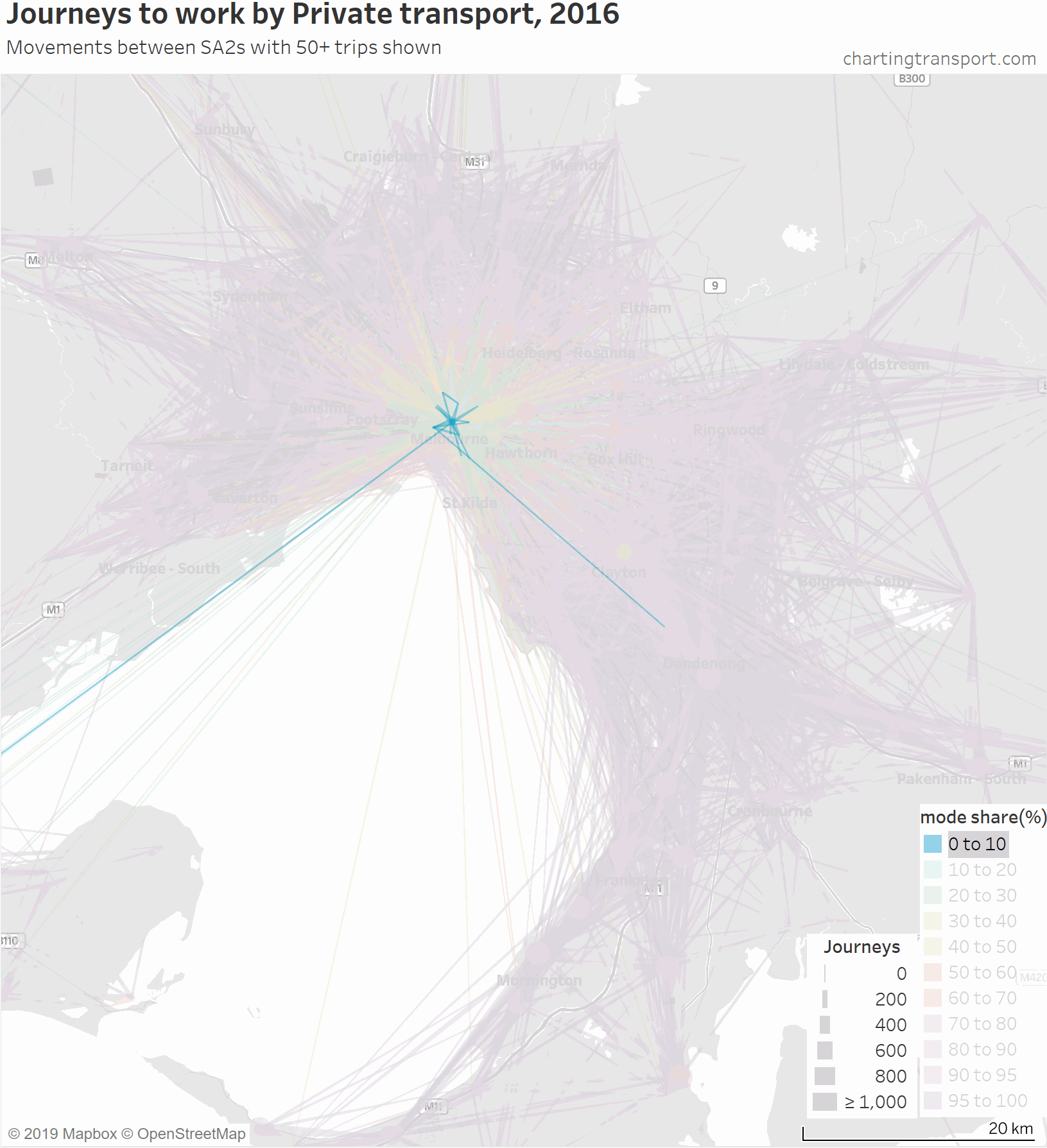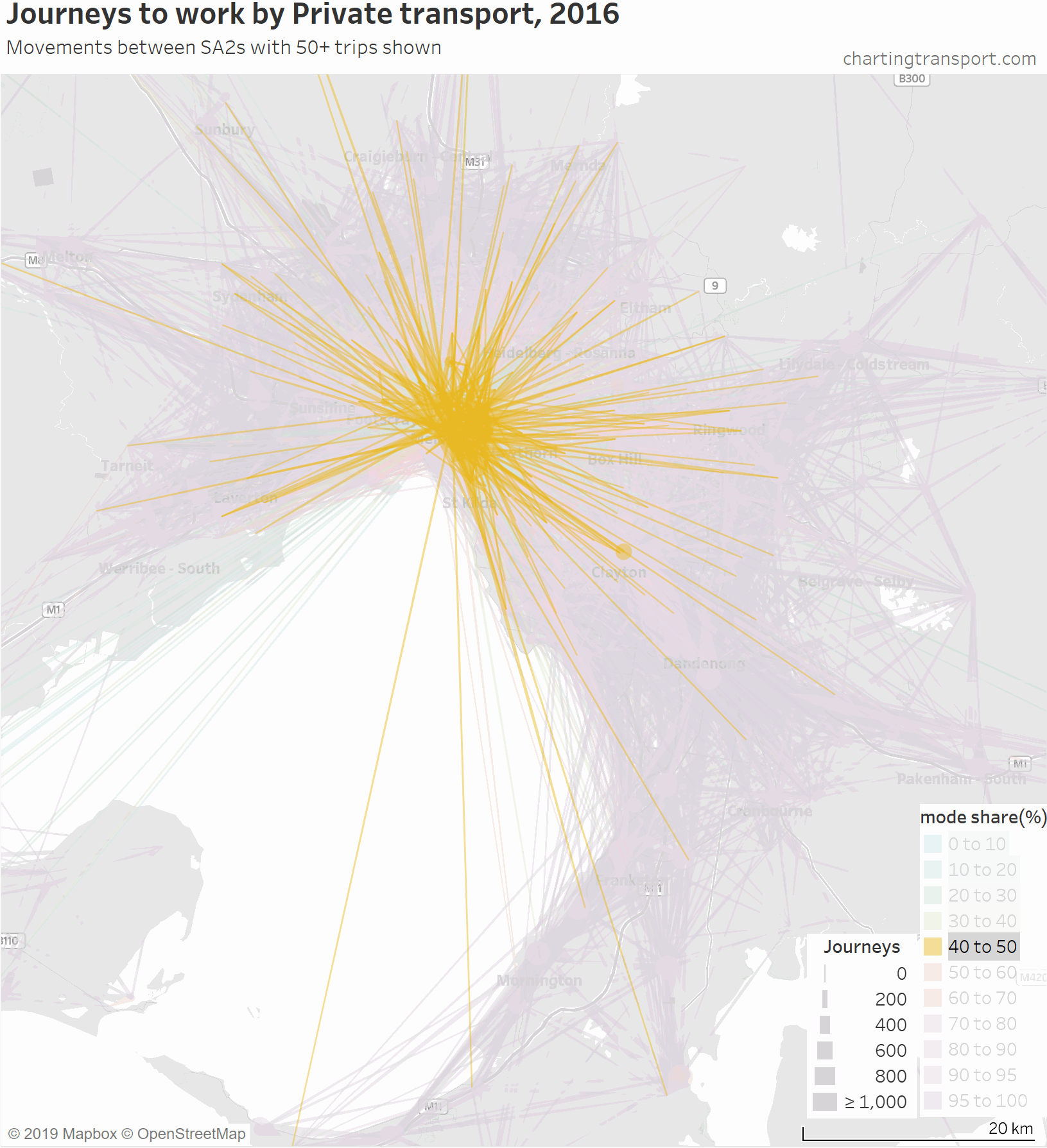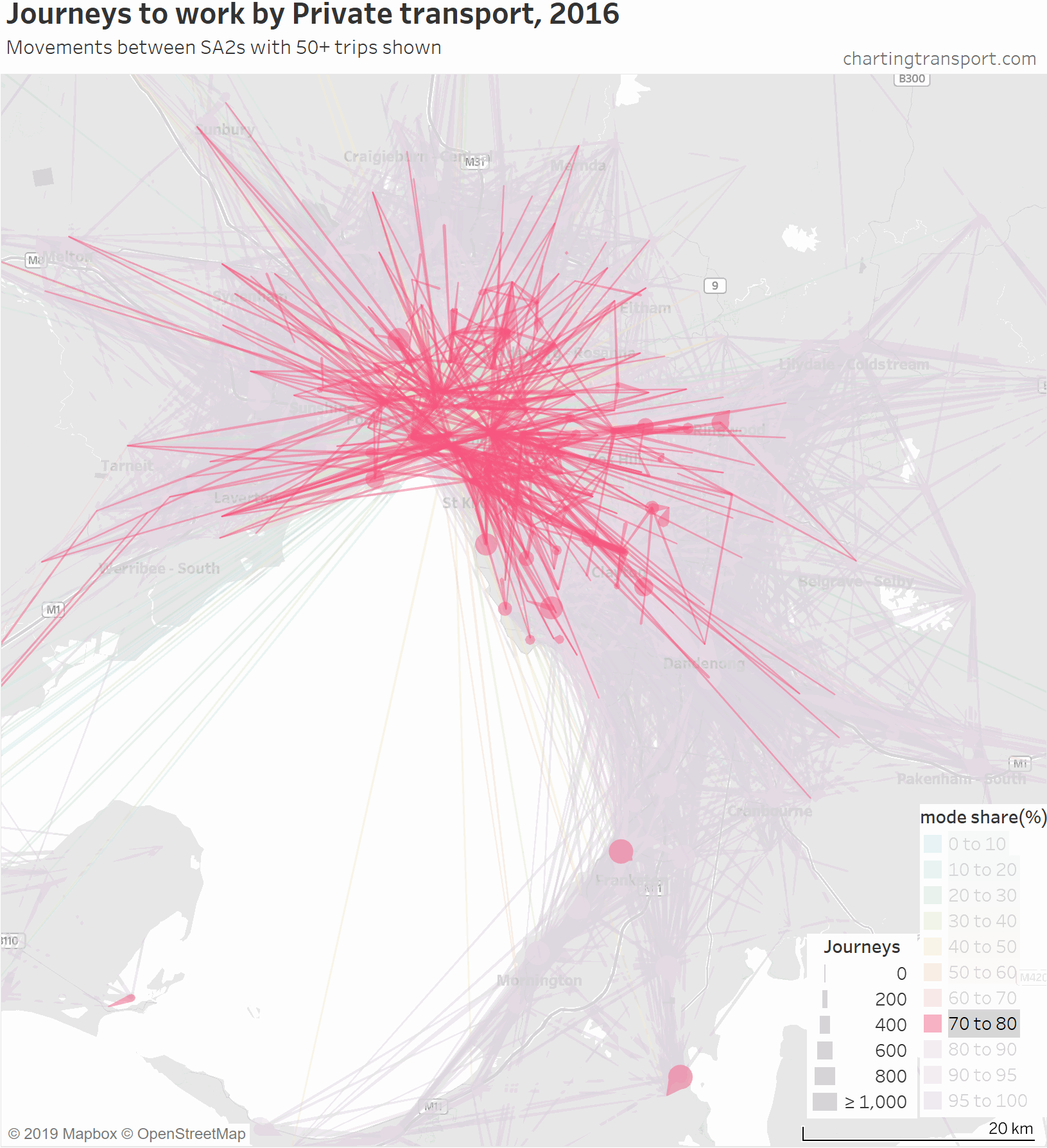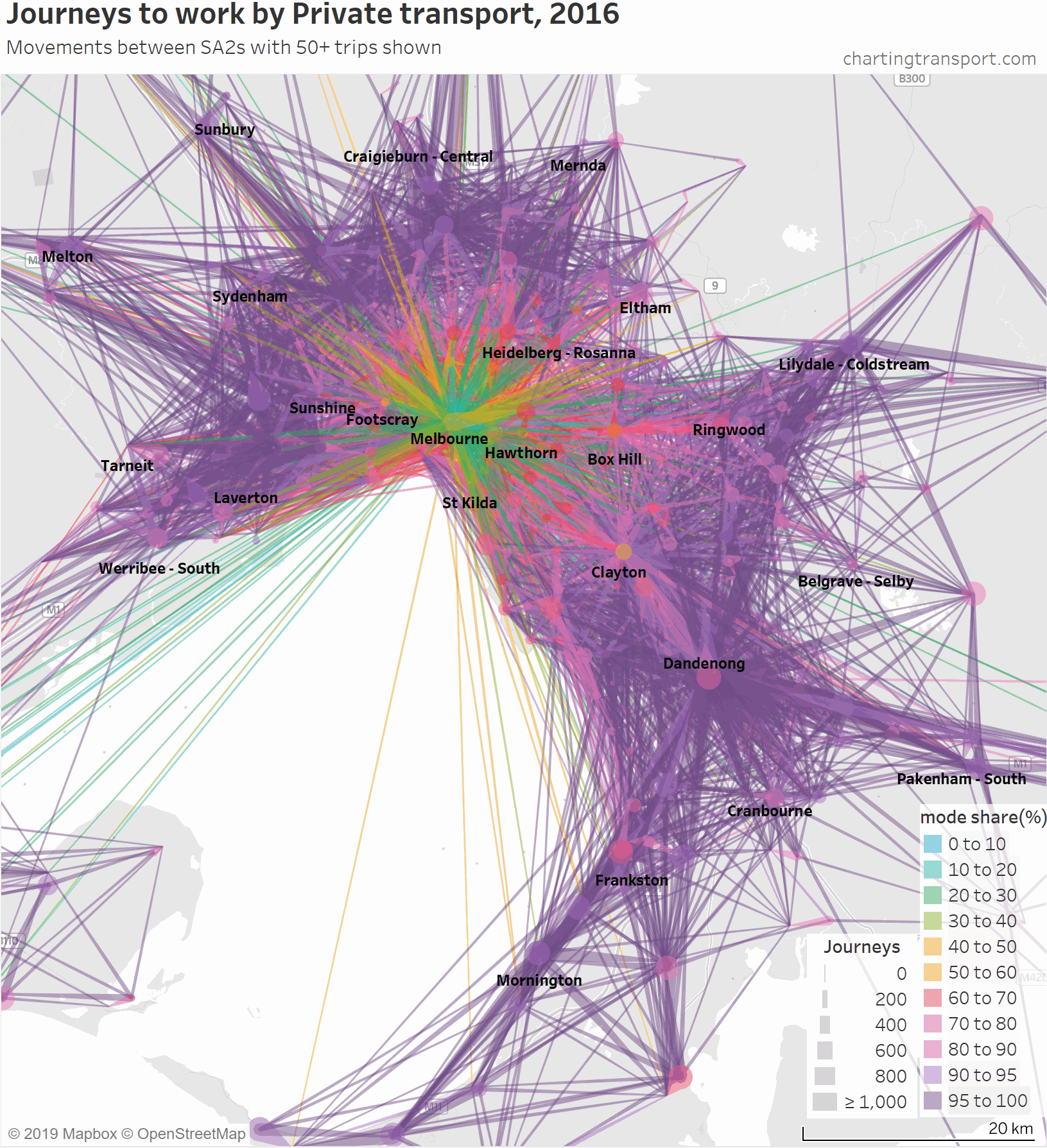
There are some other interesting stories in this data. The following map shows private transport mode share of journeys to work, excluding workplaces up to 10 km from the CBD to remove some clutter.
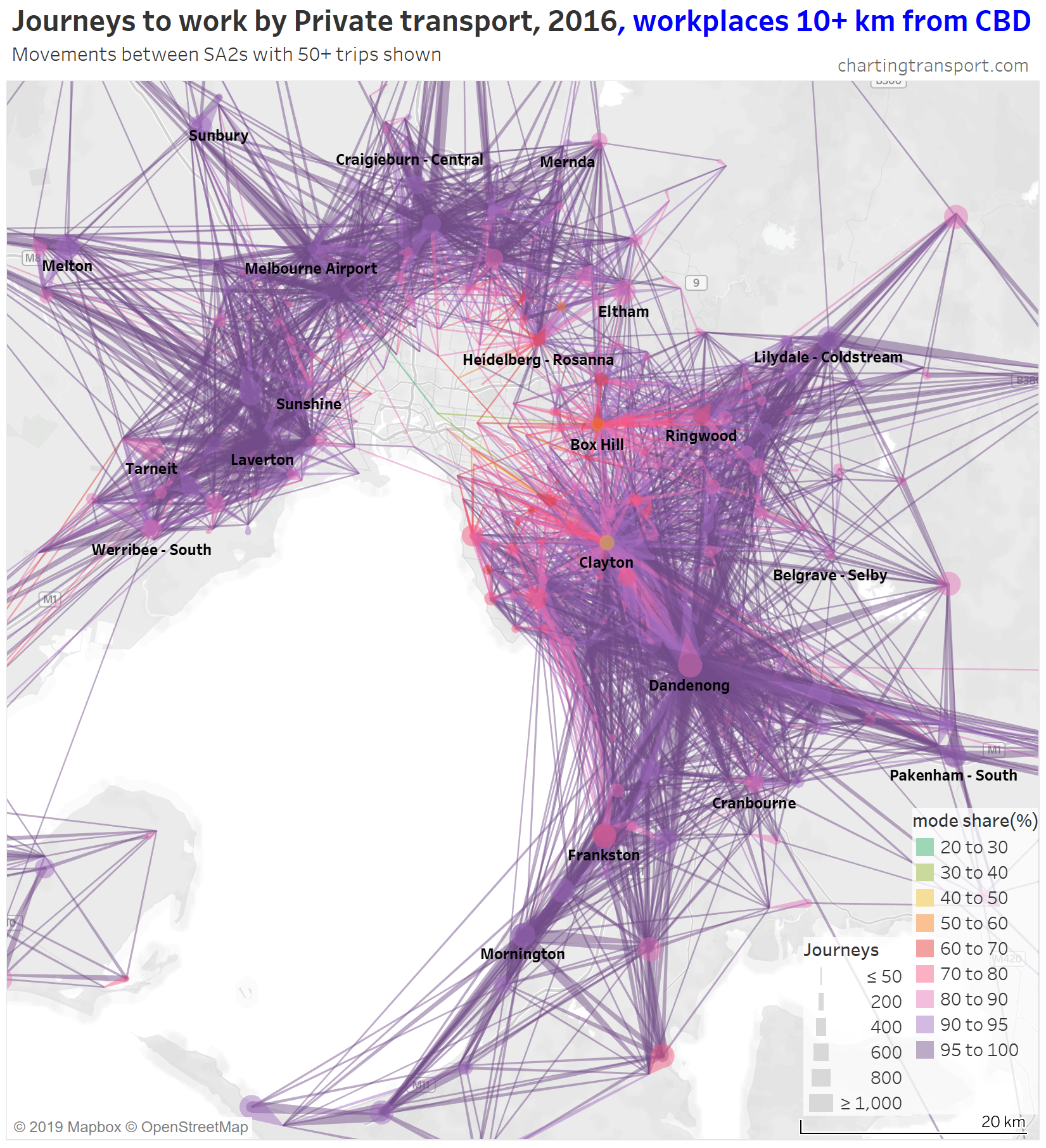
If you look carefully you’ll see that there is a much lower density of trips that cross the Yarra River (which runs just south of Heidelberg and Eltham). There are limited bridge crossings, and this is probably inhibiting people considering such journeys.
The construction of the North East Link motorway will add considerable cross-Yarra road capacity, and I suspect it may induce more private transport journeys to work across the Yarra River (although tolls will be a disincentive).
What about active transport journeys?
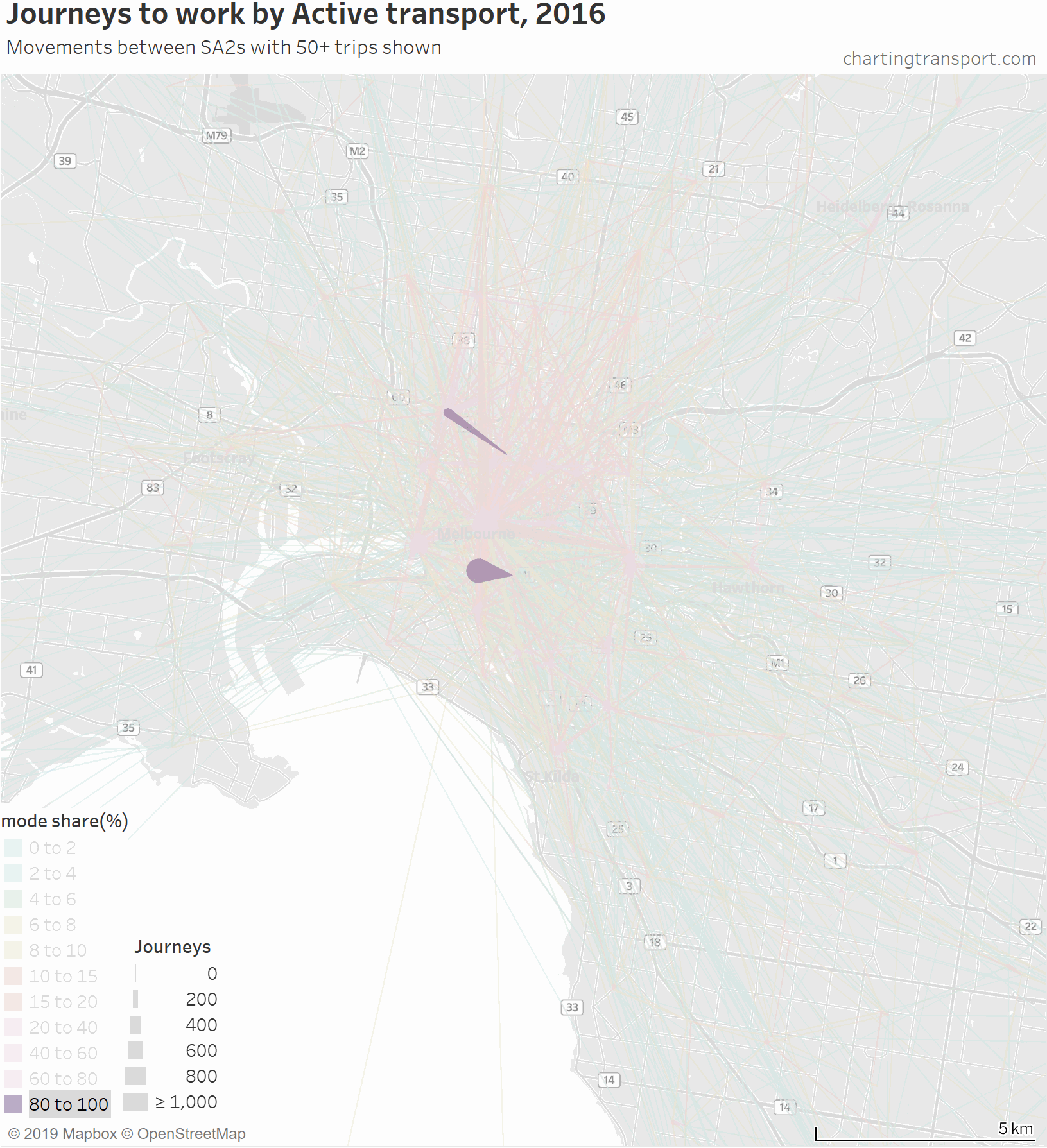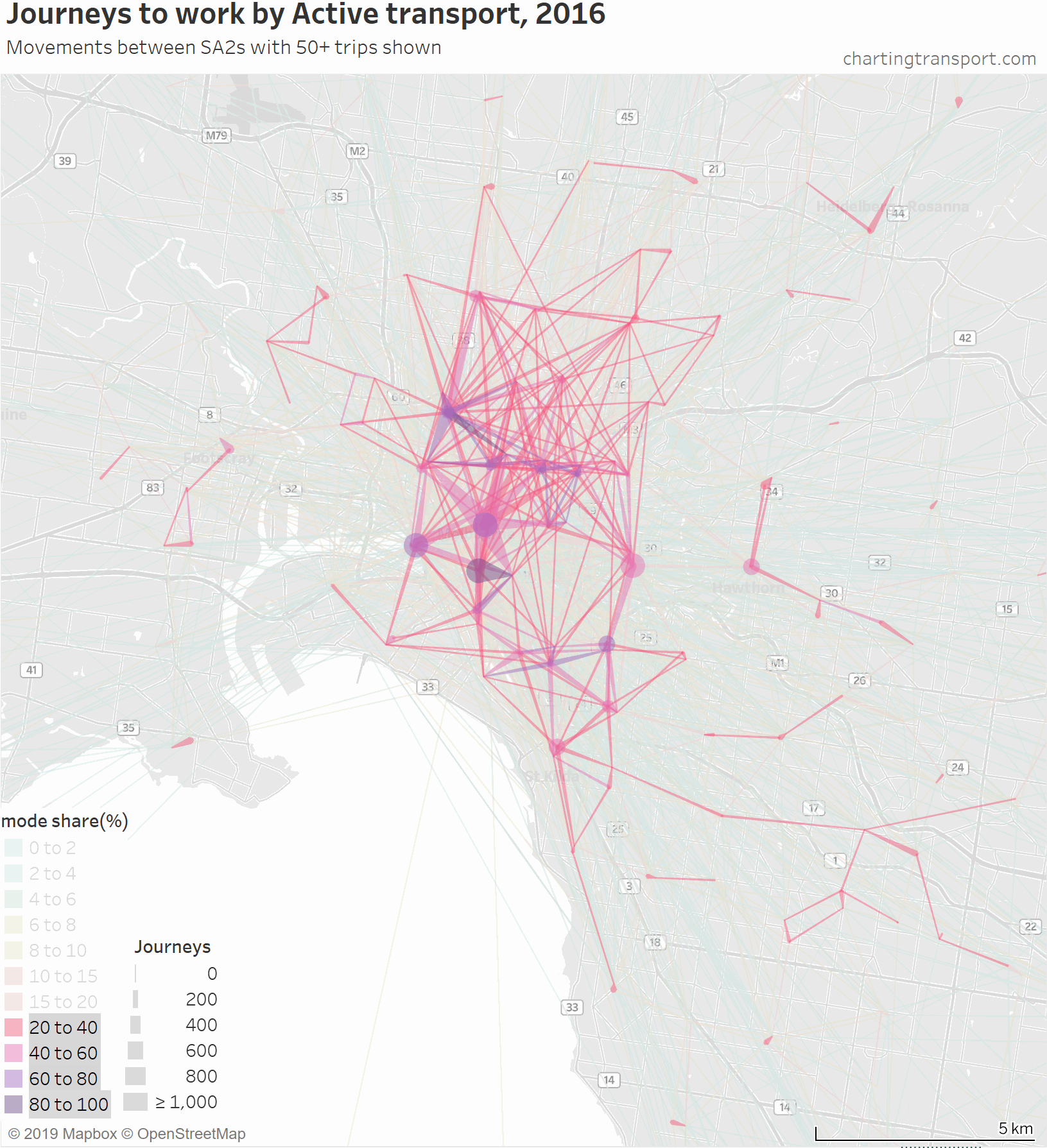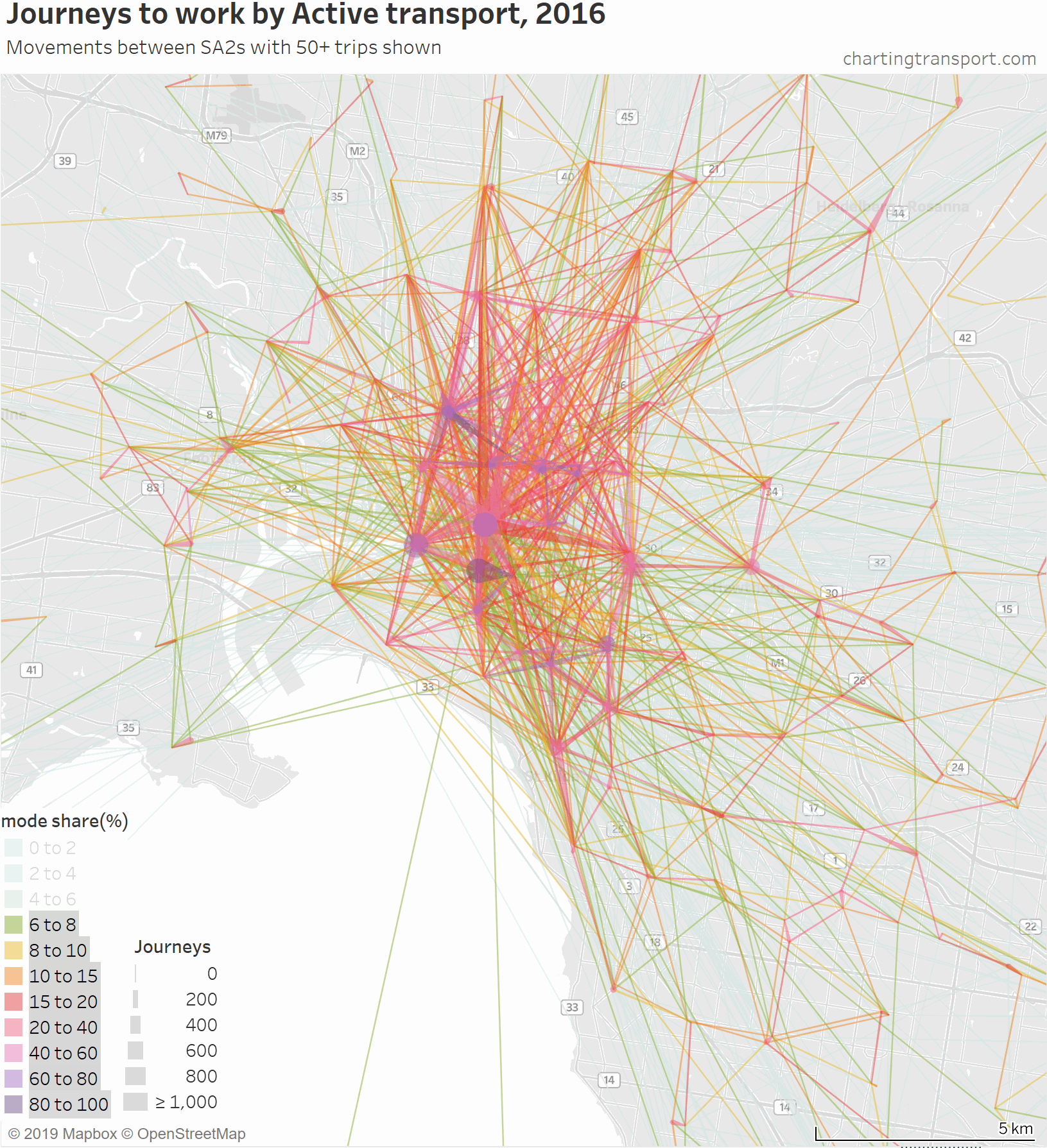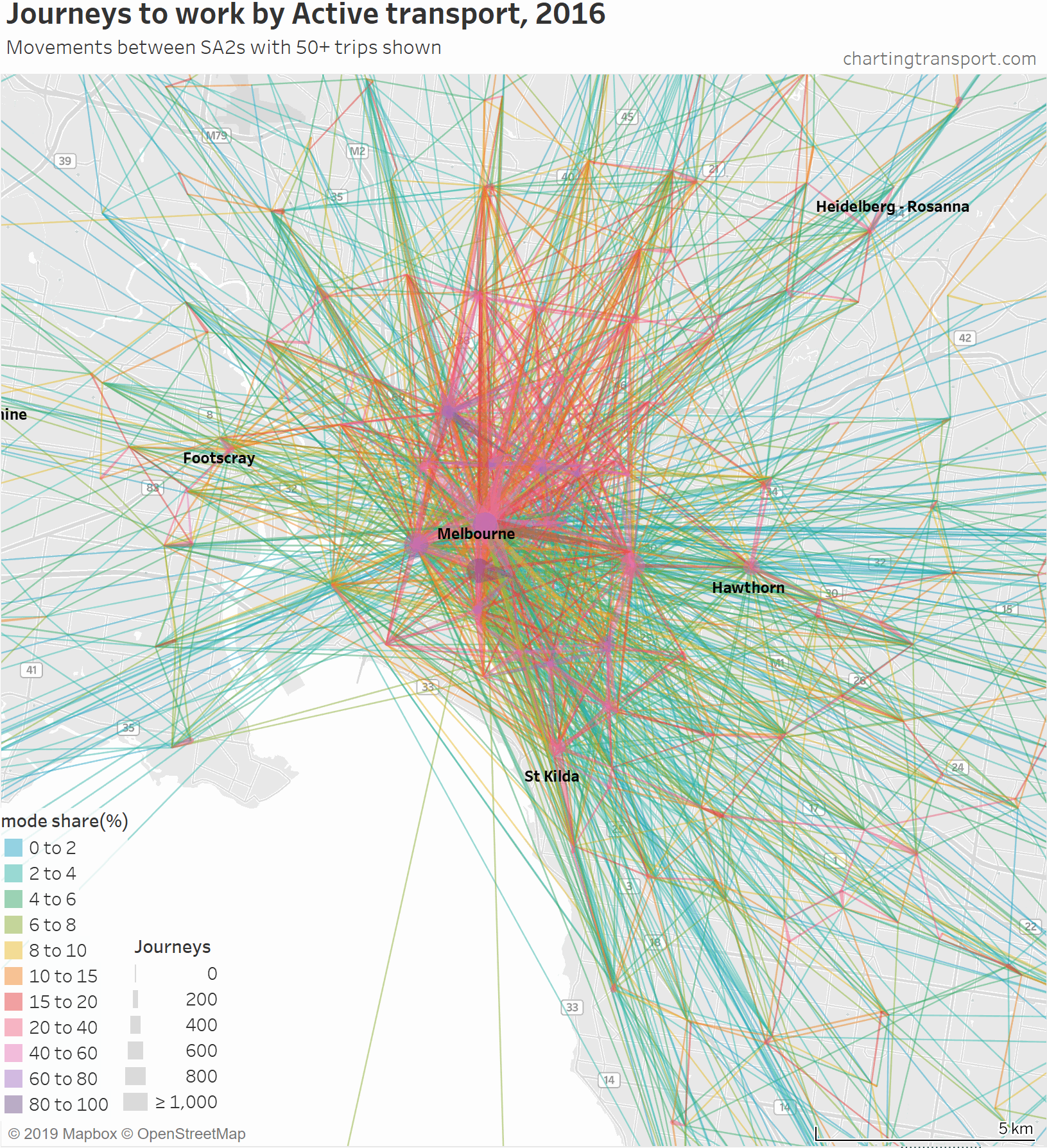
Next is a map for active transport journeys, but this time I’ve progressively added a filter for the number of active transport journeys, as most of the lines on the full chart are for very small volumes.
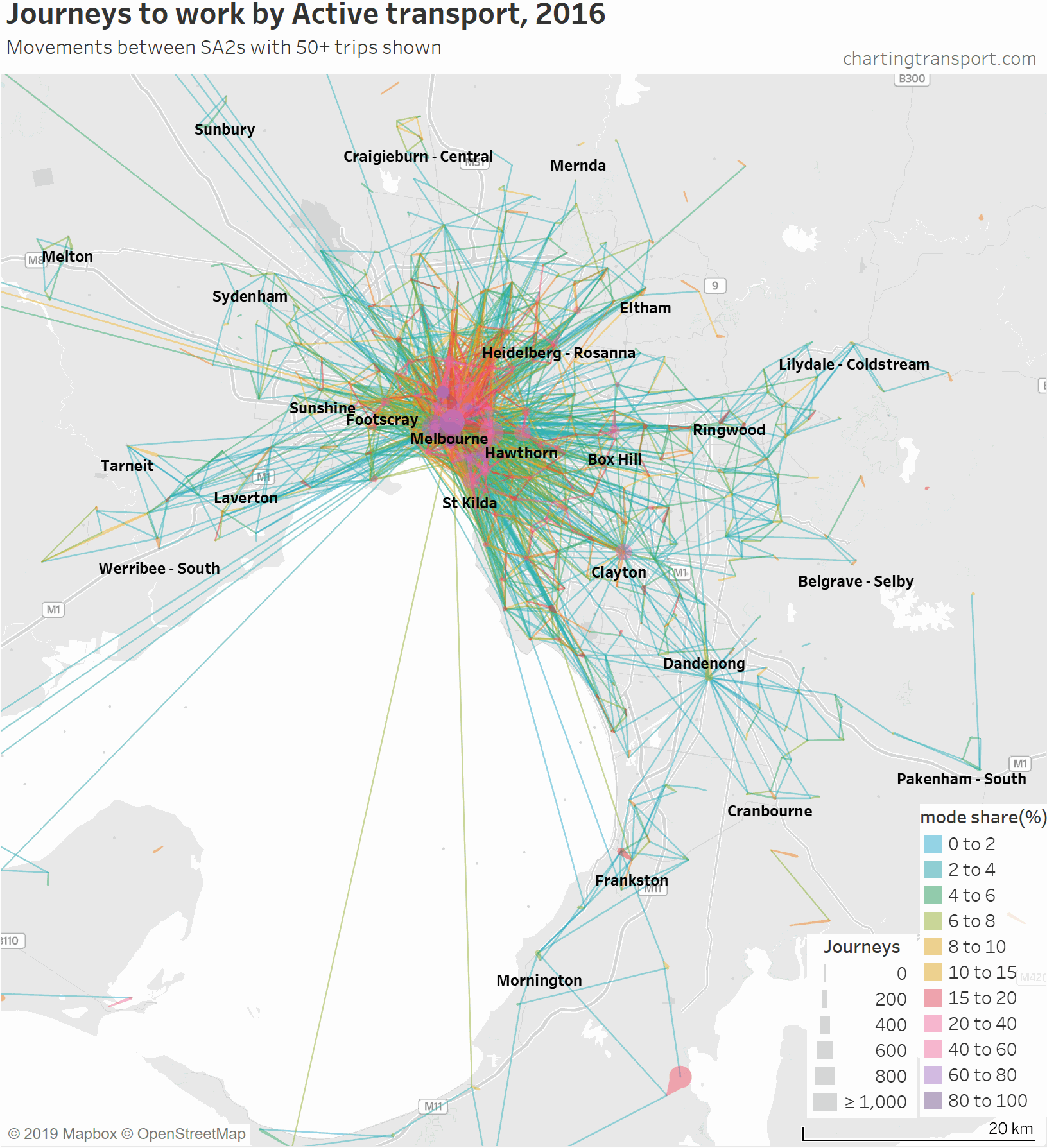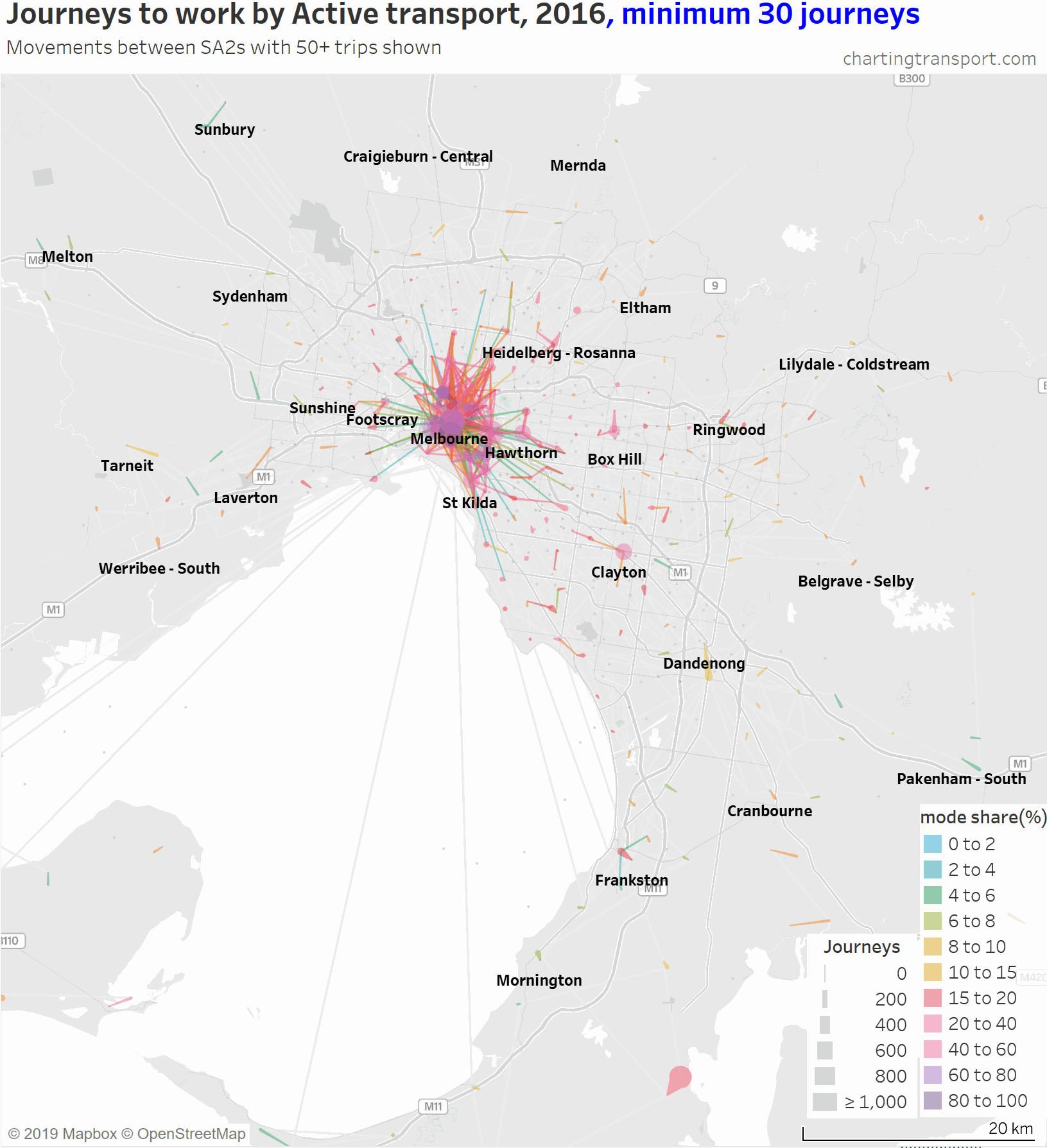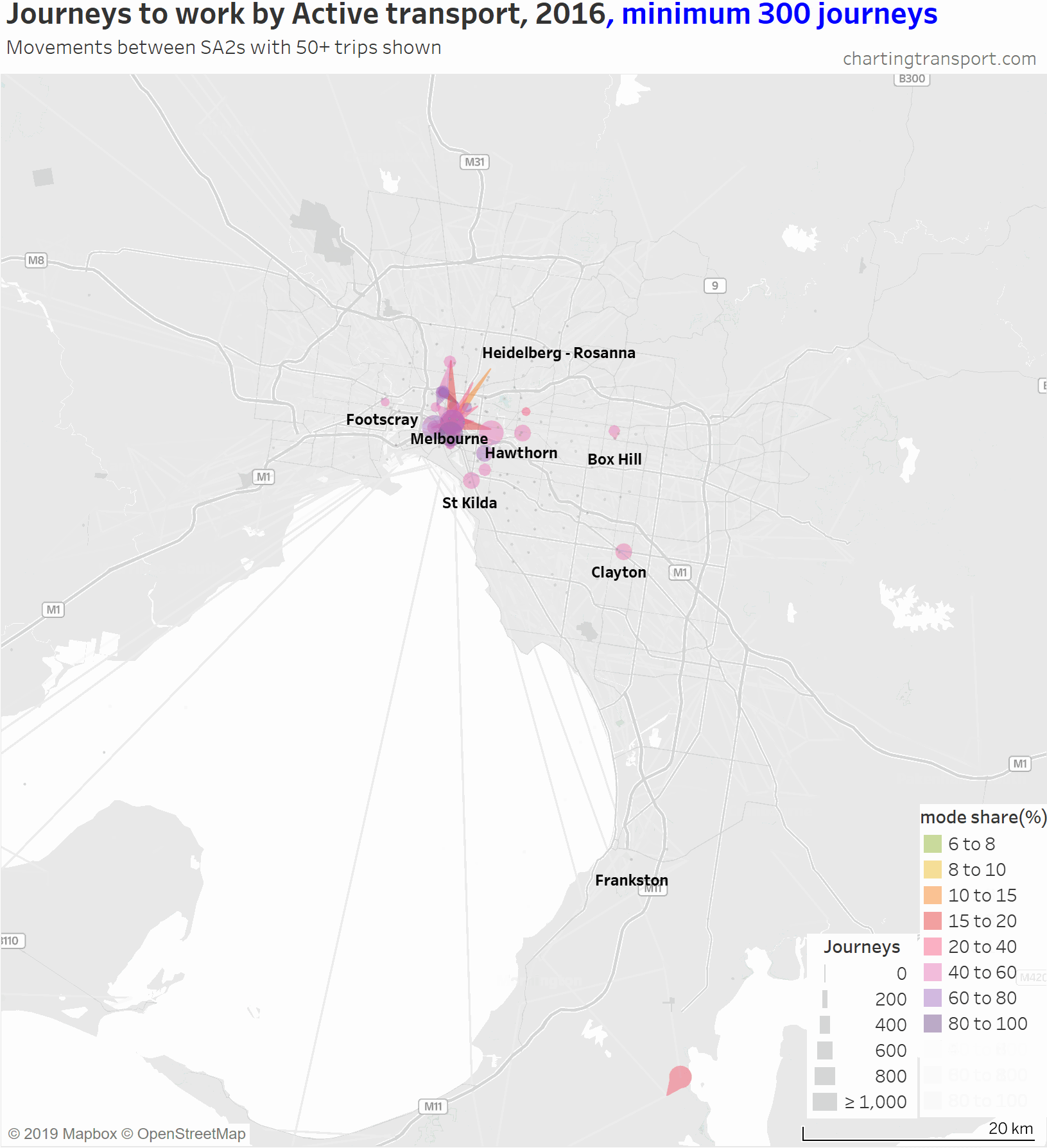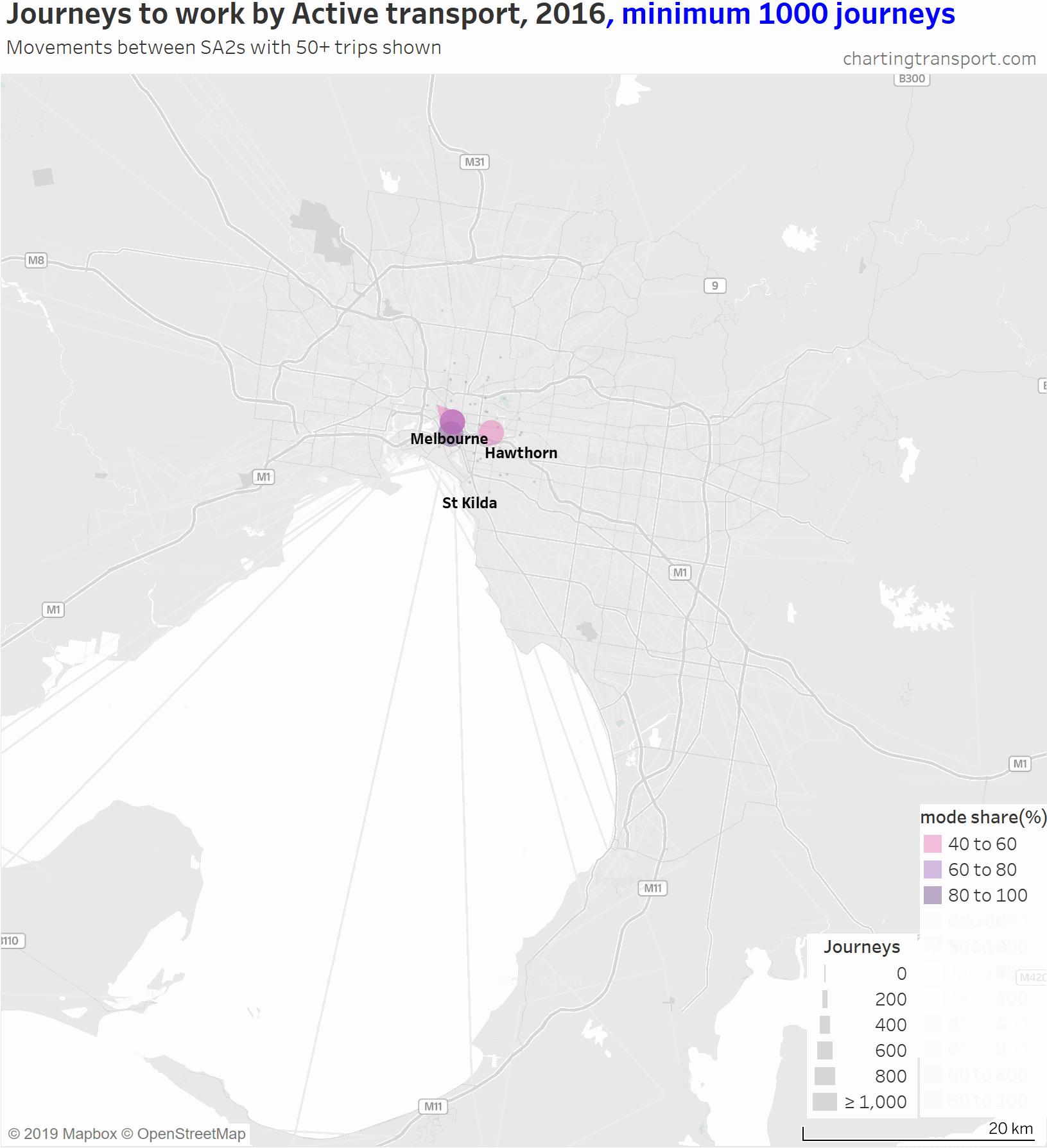
As soon as the filter reaches a minimum of 50 active journeys most of the lines between SA2s in the middle and outer suburbs disappear. Note that journeys between SA2s are not necessarily long, they might just be a short trip over the boundary.
Then at minimum 200 journeys you can only see central city journeys plus intra-SA2 journeys in relatively dense centres such as Hawthorn, Heidelberg, Box Hill, Clayton, Frankston, Mornington, Footscray, and St Kilda. The large volume in the south of the map that hangs around is Hastings – Somers, where 882 used active transport (probably mostly walking to work on the HMAS Cerebus navy base).
Active transport journeys are mostly much shorter than private and public transport journeys – as you might expect as most people will only walk or ride a bicycle so far. But there are people who said they made very long active transport journeys to work – the map shows some journeys from Point Nepean, Torquay, Ballarat, Daylesford, and Castlemaine to Melbourne. That’s some keen cyclists, incredible runners, people who changed jobs in the week of the census (the census asks for work location the prior week, and modes used on census day), and/or people who didn’t fill in their census forms accurately. The volumes of these trips are very small (mostly less than 5).
That map is very congested around the central city, so here is a map zoomed into the inner suburbs and this time animated by building up the map starting with high active transport mode share lines.
The highest active transport mode shares are for travel within Southbank and from Carlton to Parkville, followed by journeys to places like the CBD, Docklands, South Yarra, South Melbourne, Carlton, Fitzroy, Parkville, and Carlton.
Then you see a lot of trips added from the inner northern suburbs, which are connected to the central city by dare-I-say “above average” cycling infrastructure across some relatively flat terrain. In particular, a thick red line on the map is for 471 active transport journeys from Brunswick to Melbourne (CBD) with a mode share of 17%. A second thick red line is Richmond to Melbourne (CBD) being 589 journeys with 16% active mode share.
Another way of summarising mode shares by work and home distance from the CBD
I’ve experimented with another visualisation approach to overcome the clutter issues. The next charts have home distances from the CBD on the Y axis, work distances from the CBD on the X axis, bubble size representing number of journeys, and colour showing mode shares. I’m drawing smaller journey volumes on top, and I’ve used some transparency to help a little with the clutter.
Firstly here is public transport (animated to show each mode share range individually):
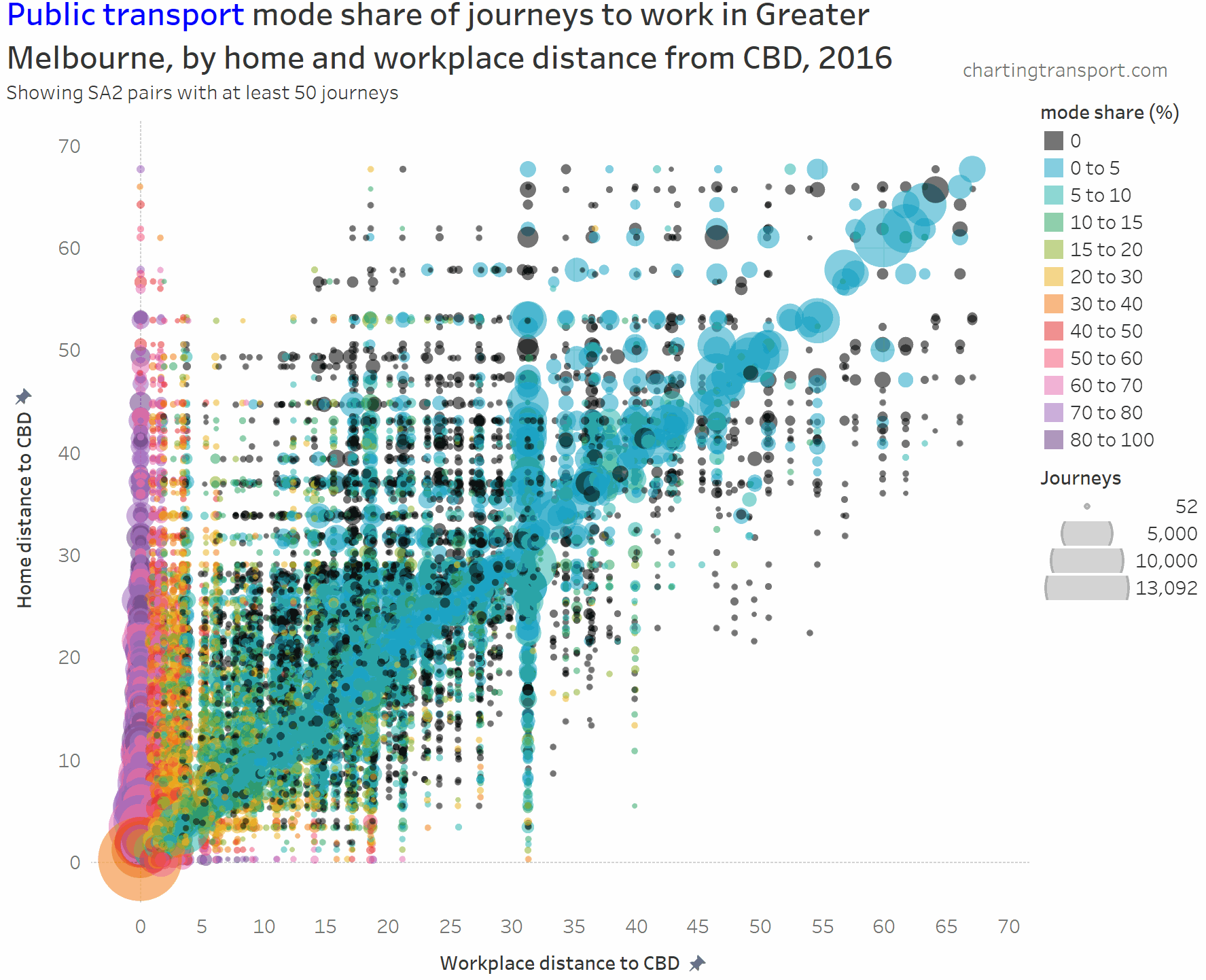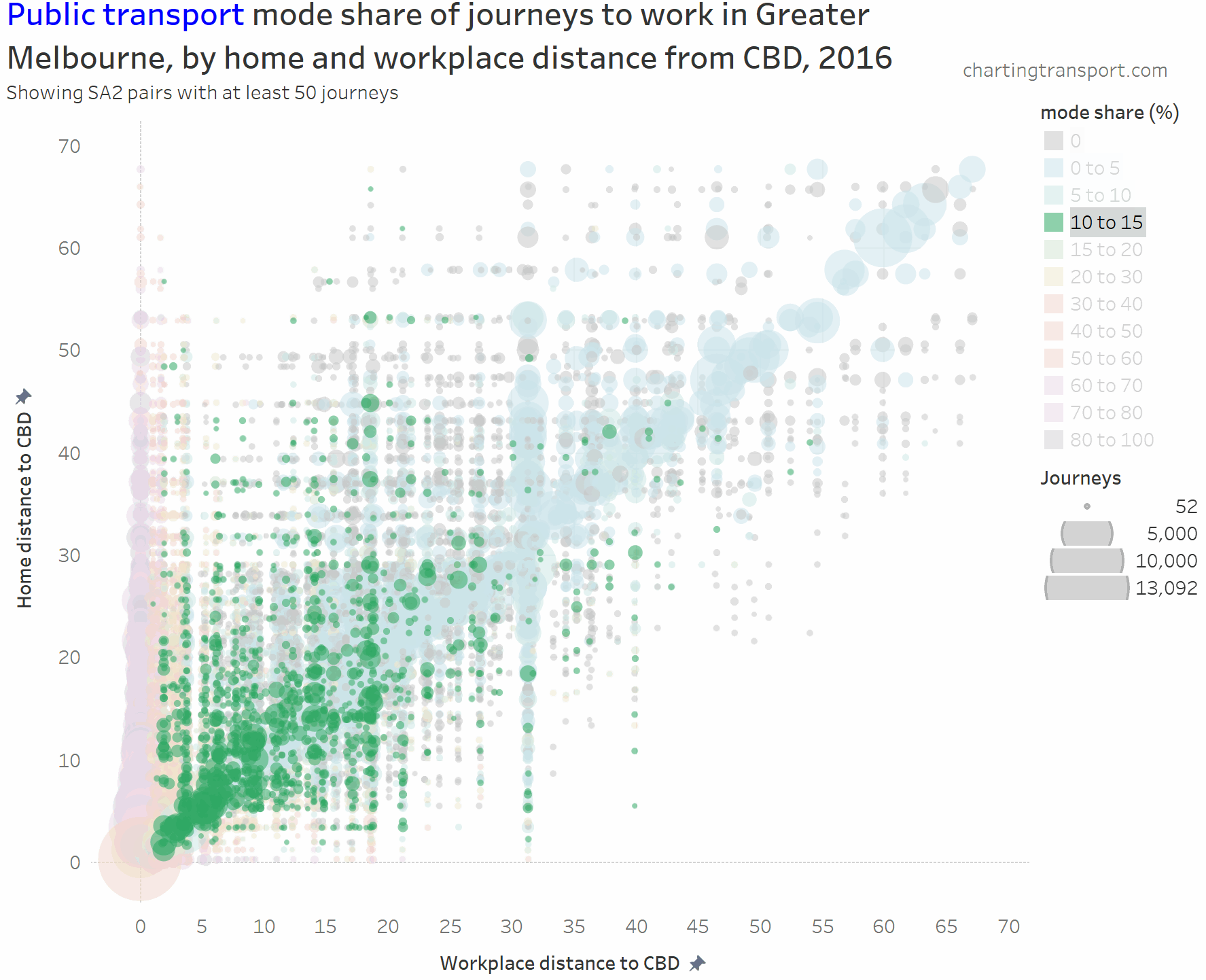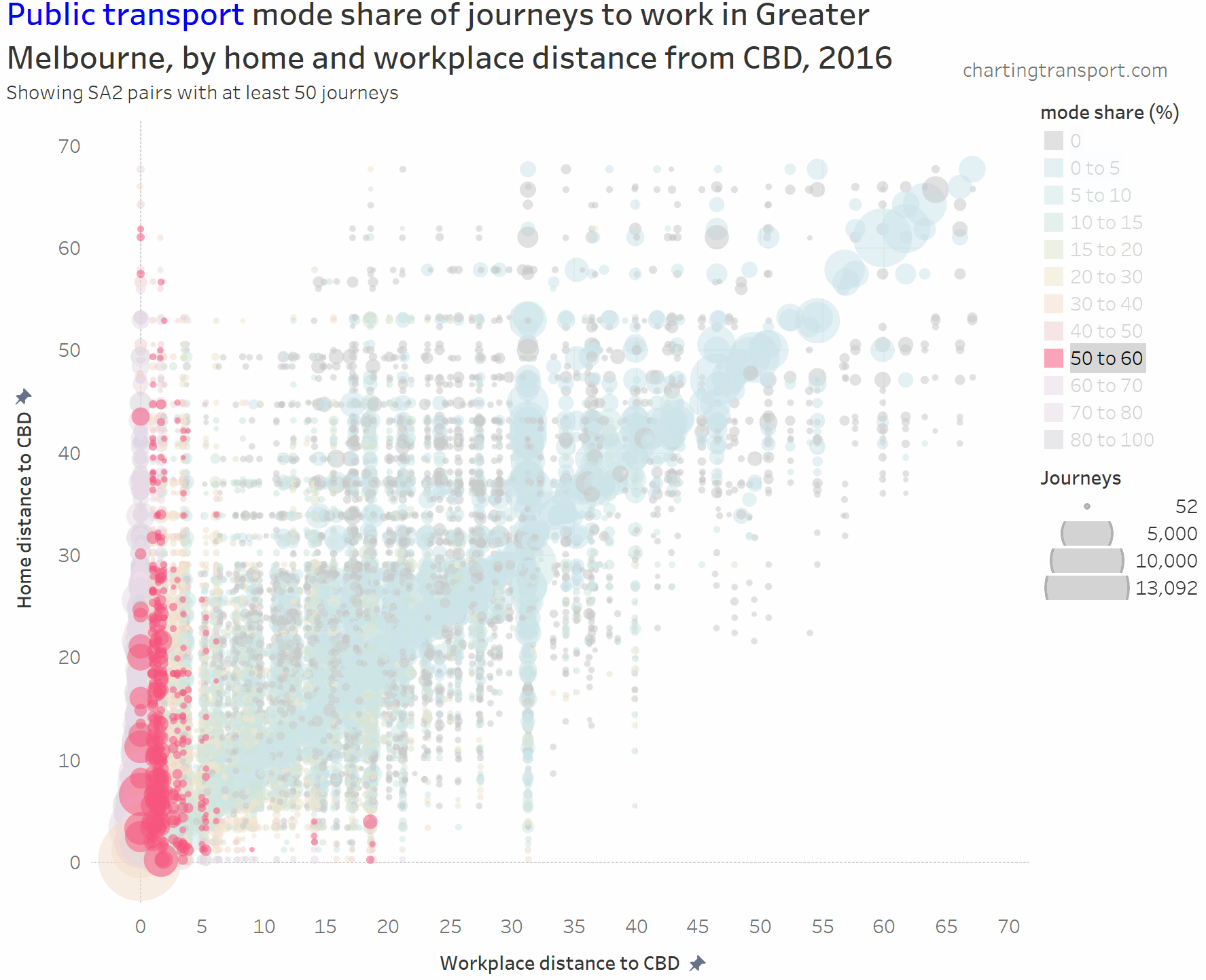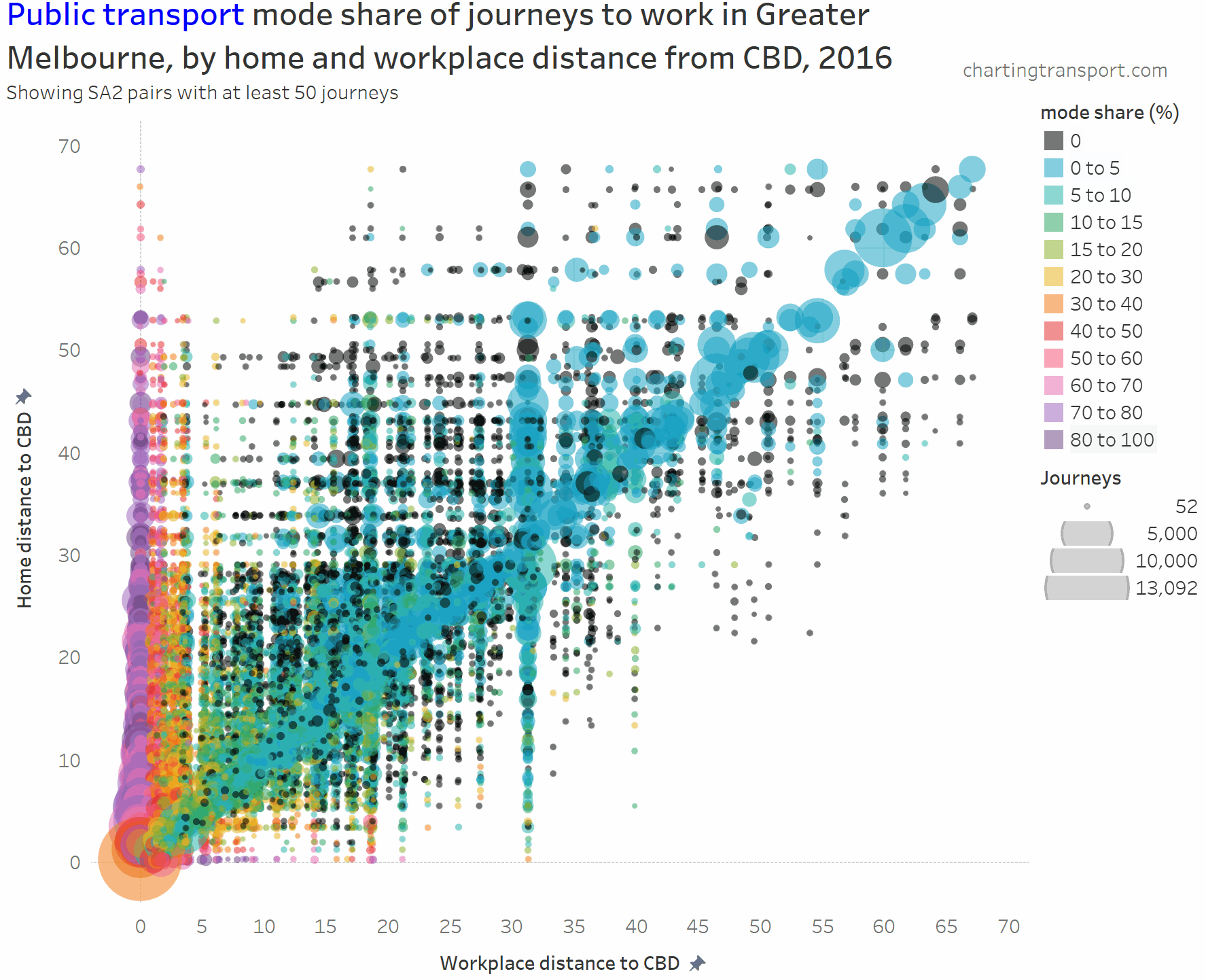
The chart is roughly a V-shape with many trips on the left edge and along a diagonal (mostly representing intra-SA2 journeys), then with several vertical stripes being major suburban employment destinations (including Dandenong at 31 km, Clayton at 19 km, and Frankston at 40 km). Trips above the diagonal are roughly inbound, while trips below the diagonal are roughly outbound.
Some observations:
- The diagonal line (mostly local journeys) has very low public transport mode shares (sometimes zero).
- Higher PT mode shares are only seen on the far left and bottom left hand corner of the chart. Some outliers include Richmond to Box Hill (32%), Clayton to Malvern East (32%), and South Yarra – East to Clayton (57%).
- PT mode shares of 80+% are only seen for journeys to the CBD from home SA2s at least 11 km out (with one exception of Melbourne CBD to St Kilda with 80% PT share).
- Home-work pairs with zero public transport journeys are scattered around the middle and outer suburbs, most being longer distance journeys (home and work at different distances from the CBD).
Here’s the same chart for private transport:
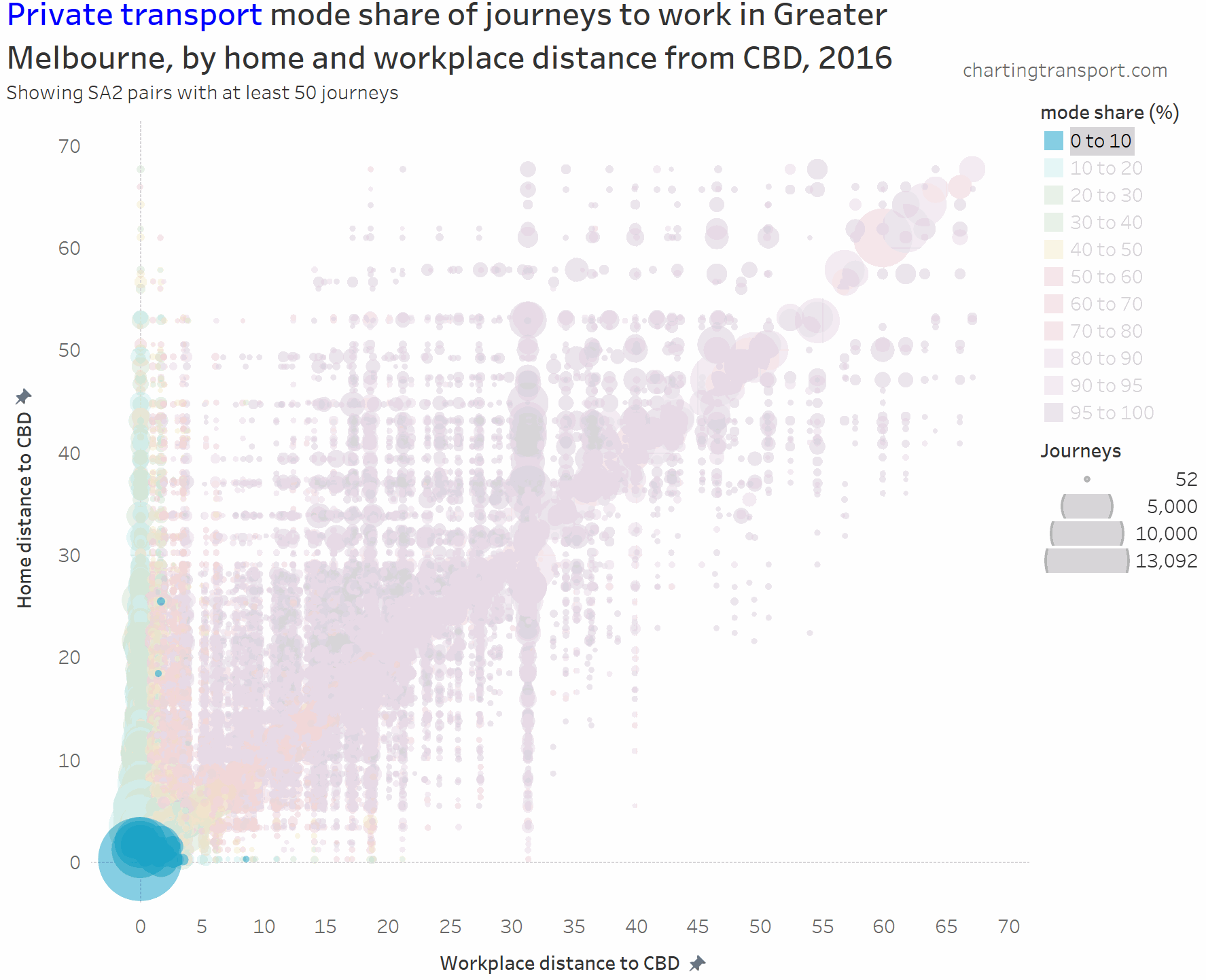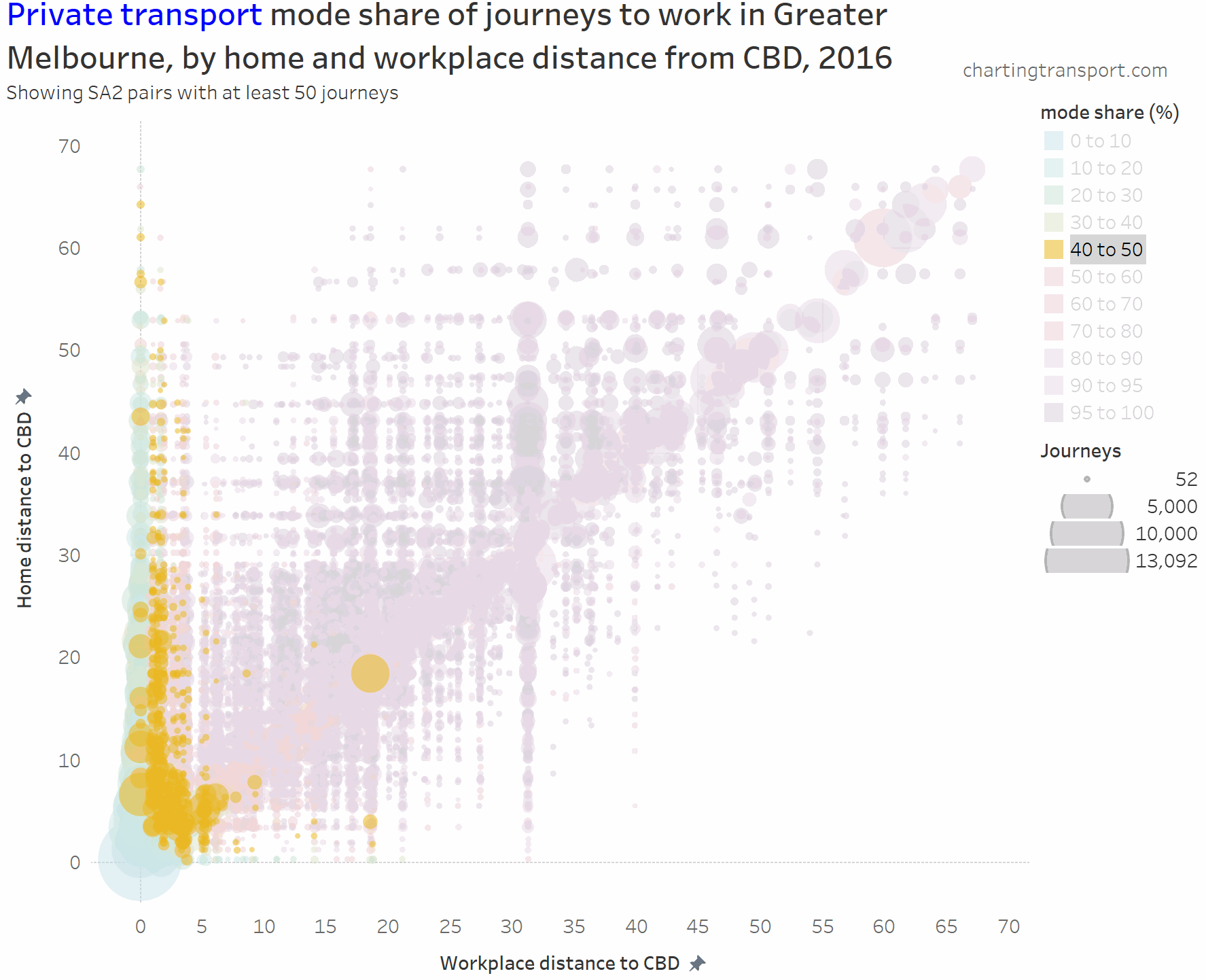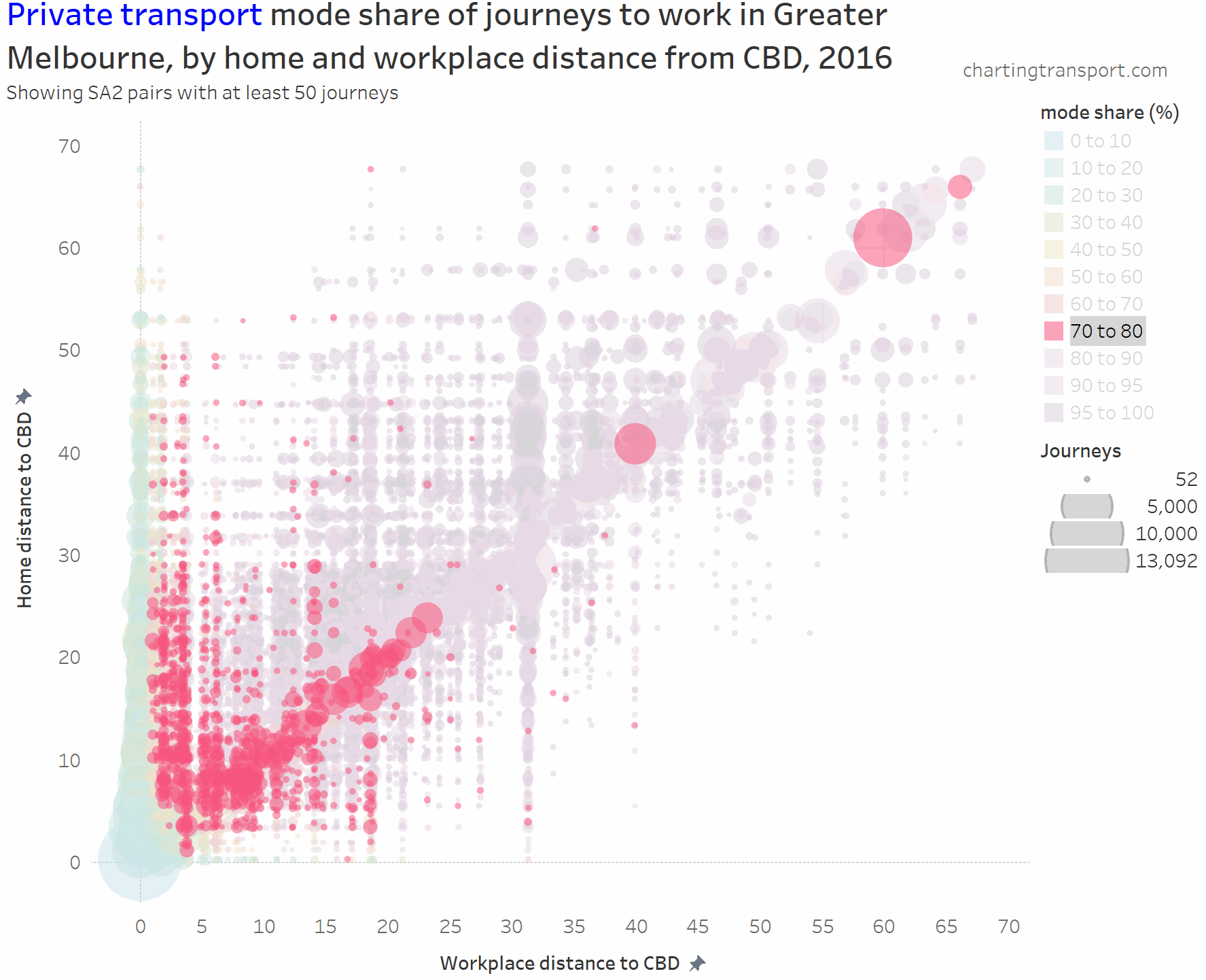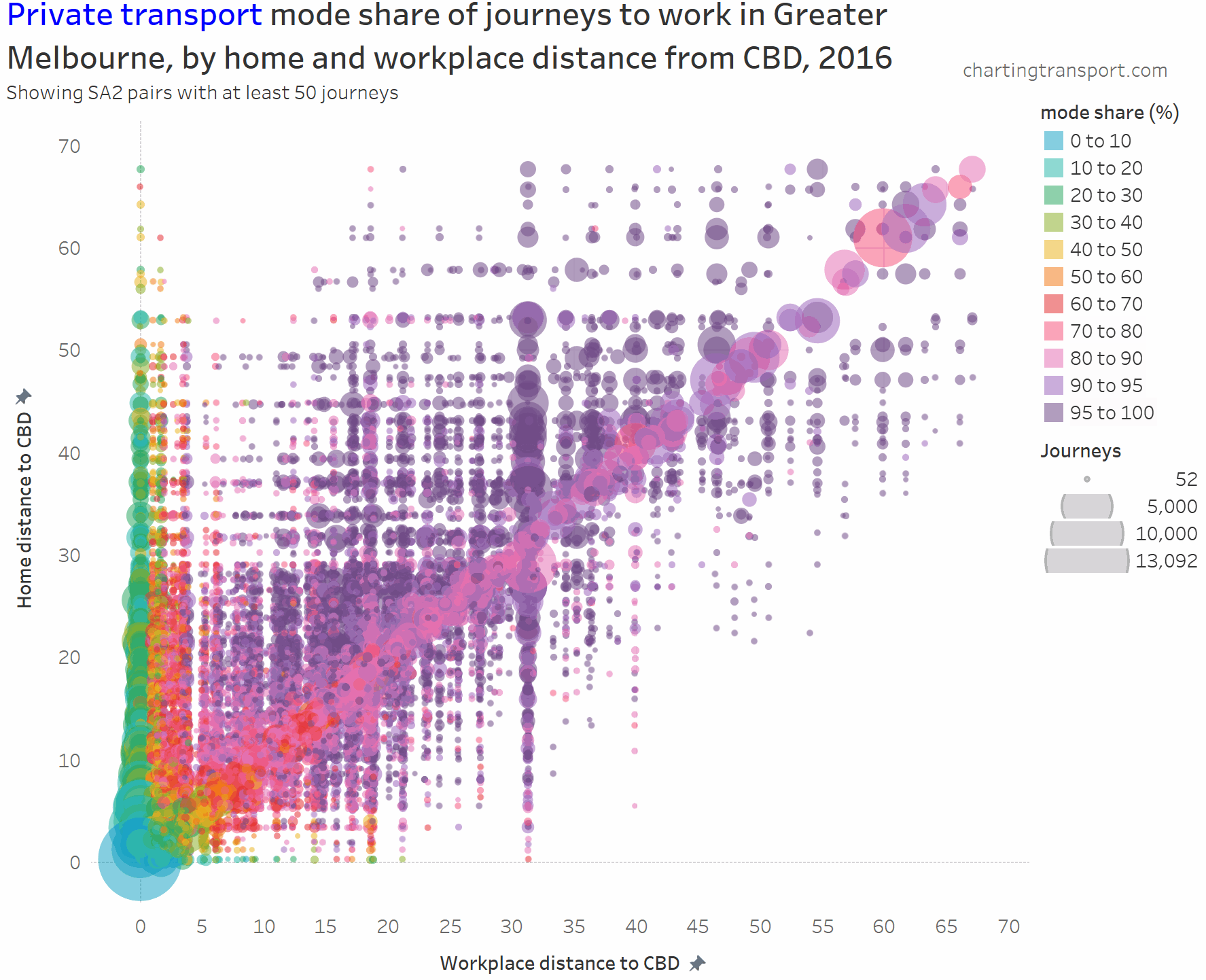
The lowest private transport shares are seen for journeys to the CBD. The diagonal has many mode shares in the 80-90% range.
And here is active transport:
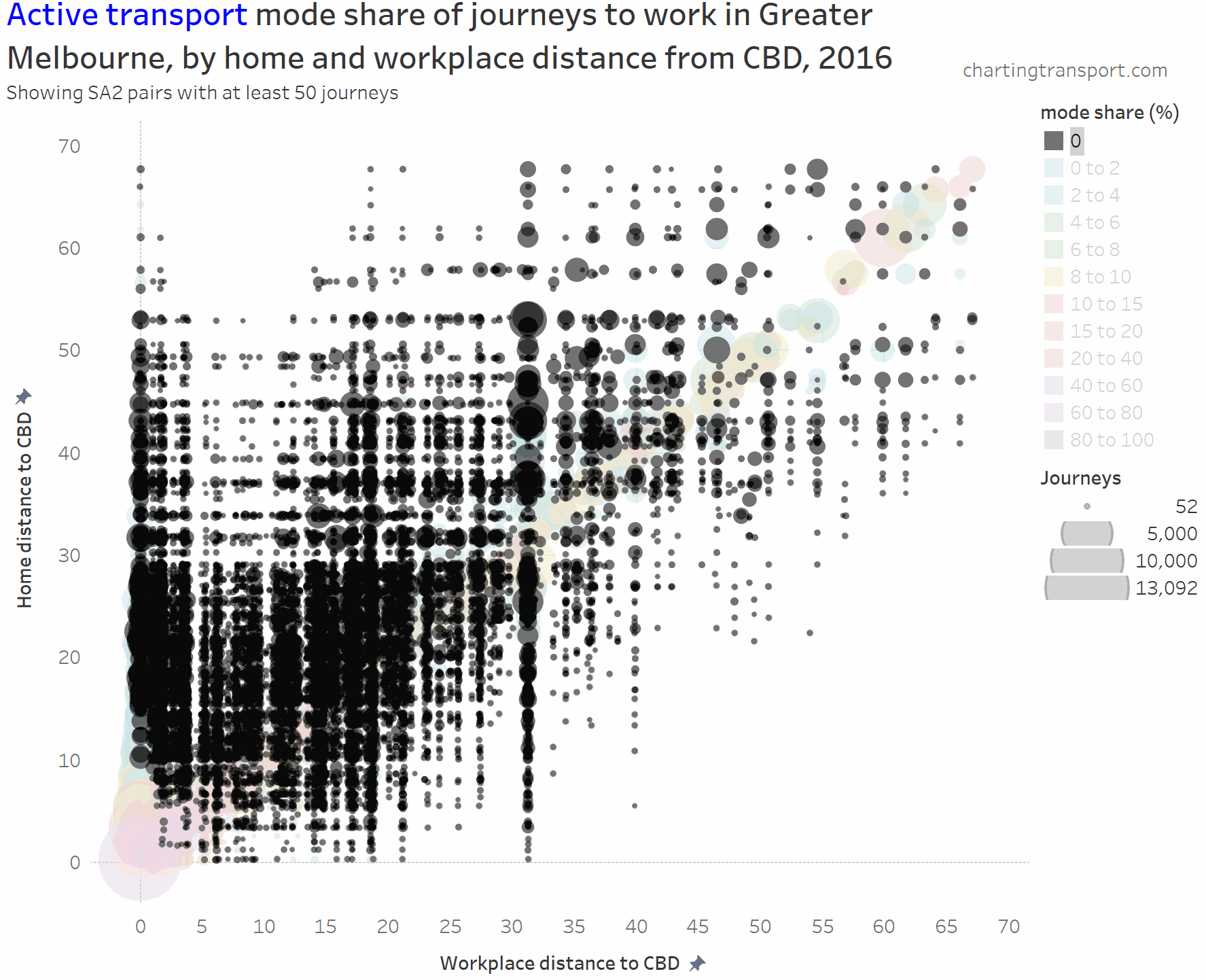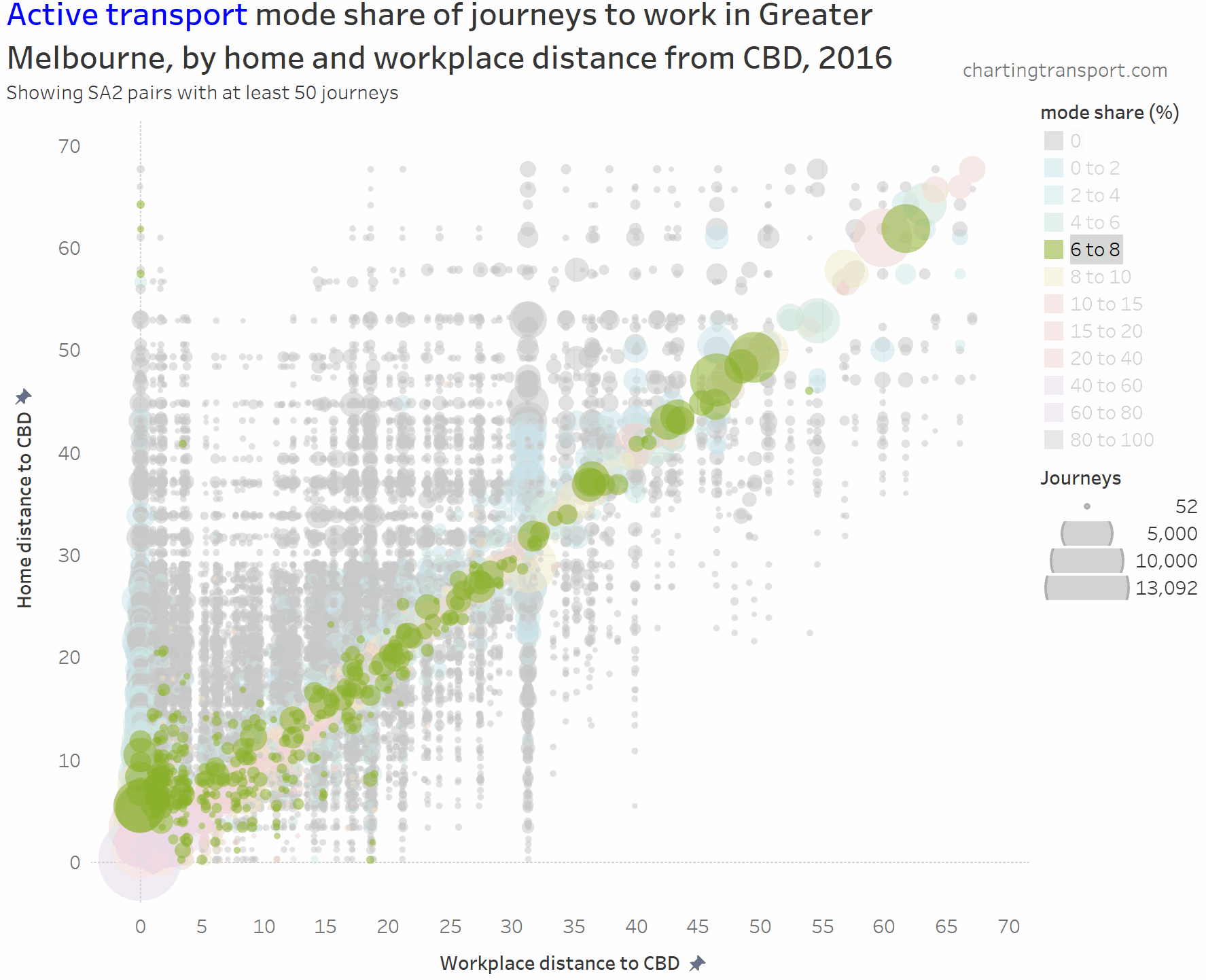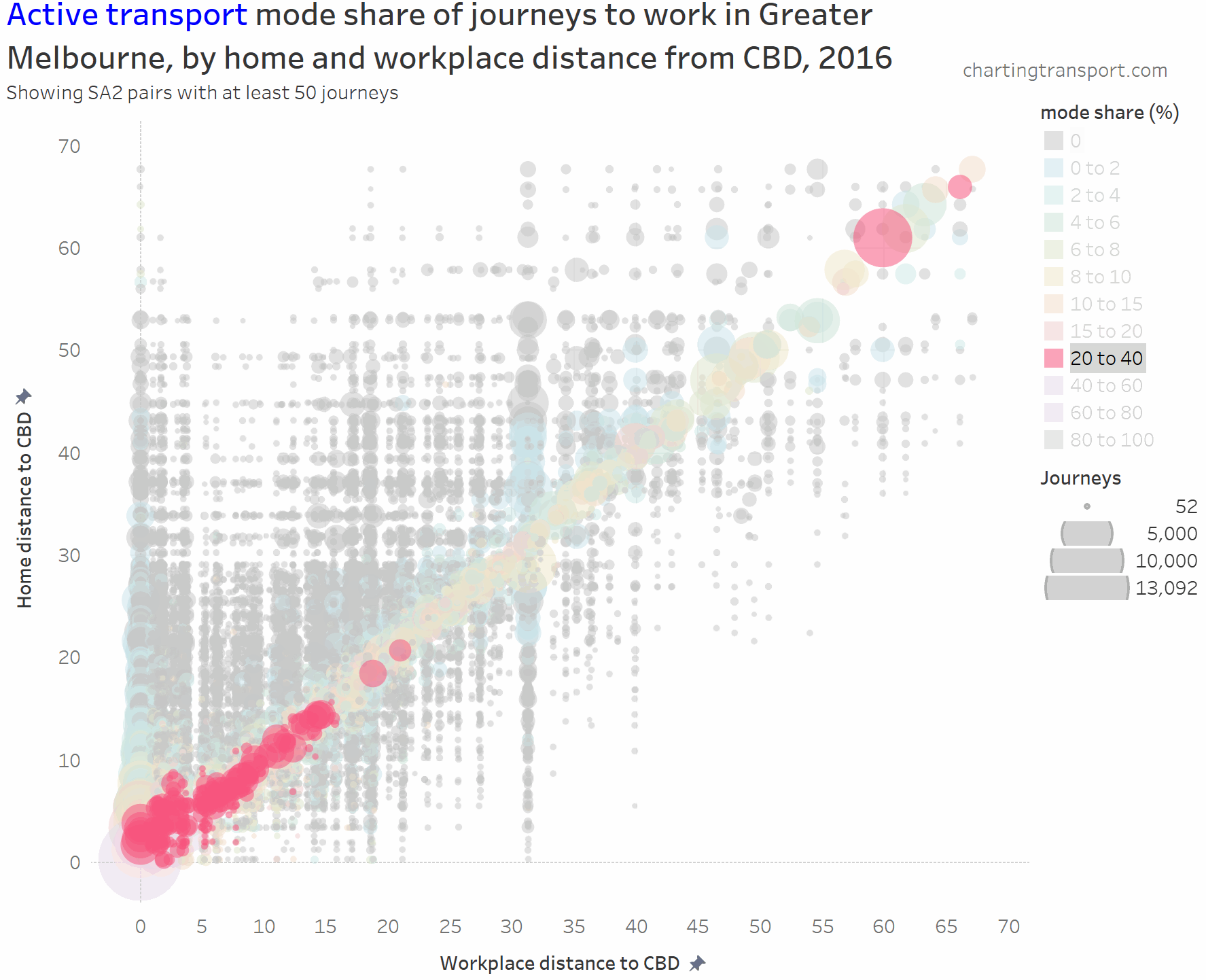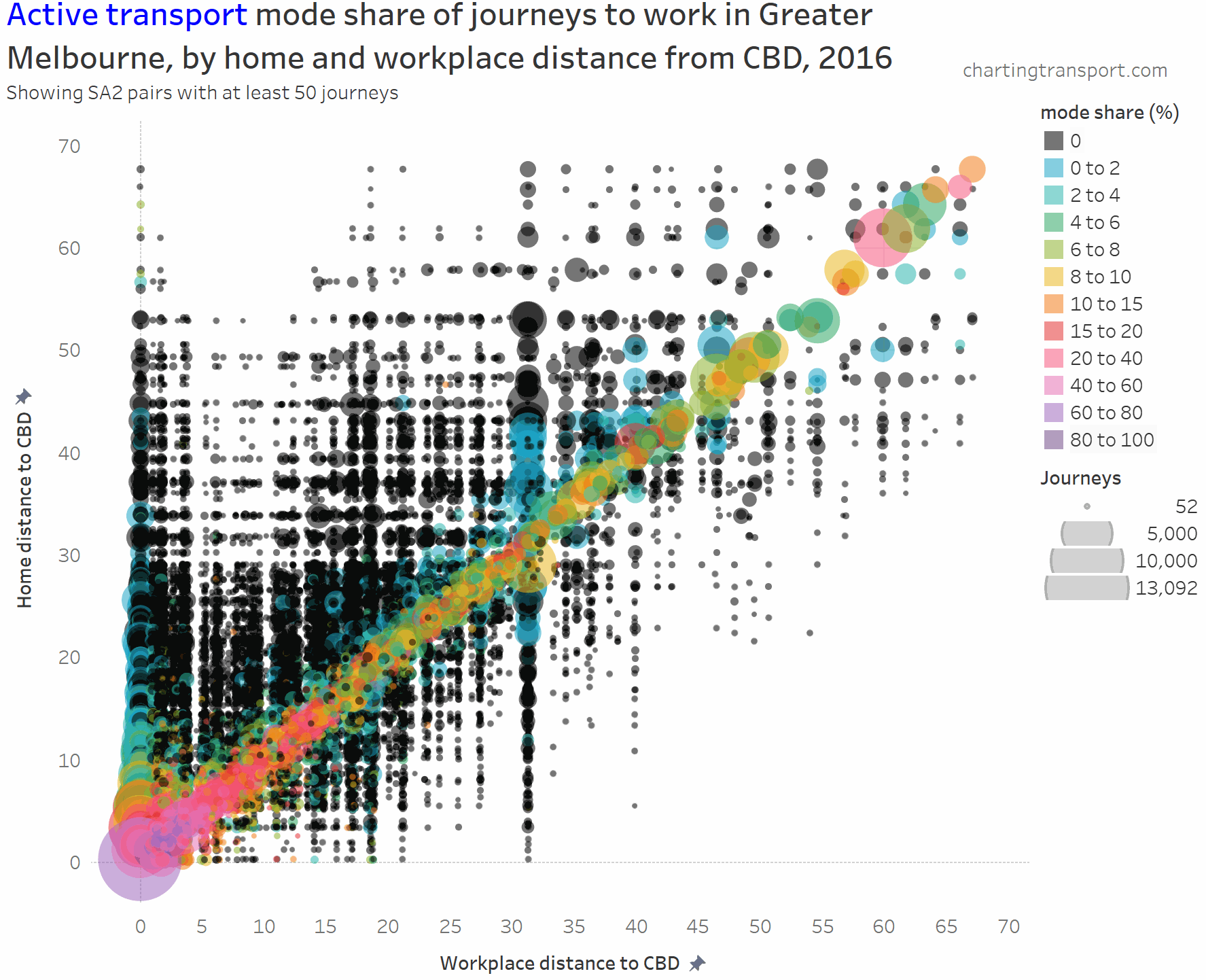
The highest active transport mode shares are seen in the central city area, followed by the diagonal mostly representing local journeys (with generally higher shares closer to the CBD). Some notable outliers include local trips within Clayton (1,298 active trips / 46% active mode share), Box Hill (914 / 40%), Hastings – Somers (1,762 / 27%), Flinders (240 / 24%), Glen Waverley – West (308 / 21%), and Mentone (226 / 23%).
How does Sydney compare to Melbourne?
Here is a chart with private transport mode share maps for both Melbourne and Sydney, animated in tandem to progressively add higher mode share journeys.

You can see that Sydney has a lot more trips at lower private transport mode shares, and that workplaces outside the city centre start to show up earlier in the animation in Sydney – being the dense transit-orientated suburban employment clusters that are largely unique to Sydney (see: Suburban employment clusters and the journey to work in Australian cities).
If time permits, I may do similar analysis for Sydney and other cities in future posts.


Chris, my own take on the low density of trips crossing the Yarra is that JTW data is dominated by short-distance travel, and there are just fewer short-distance travel opportunities that cross from one side of the Yarra to another. So less the Yarra acting as an actual barrier, than there just not being many homes or workplaces a short distance away. Conversely, the highest densities of trips occur where there’s lots of contiguous urban development.
And of course the North East Link isn’t really designed to cater for short-distance trips either.
LikeLiked by 2 people
Really interesting, and Tony, I like your thinking.
I found the work//home distance scatter plots most interesting – the fact that they’re predominantly upper diagonal also just shows most people live farther from the CBD than they work. And the animations really work well to pull the data apart.
One negative point presentation-wise though – I assume it would take more time but would you consider having the images clickable to open to their full size? I was right-clicking to open them in a separate tab, but they still had the “?w=450” suffix that I had to remove to appreciate the fine detail (and legend text) that you’ve done.
(It also enabled me to pick up one typo in journey-length-v-pt-mode-share-v-dist-from-cbd.png with the website being “chartingtranpsortcom”)
LikeLike
I think Chris’ take is right – the lack of Yarra crossings simply means people’s geographic networks rarely cross the Yarra as you’ve got Chandler Hwy, Fitzsimons Lane, and Warrandyte bridge. That’s it. Not exactly the arterial grid of the eastern suburbs that allows many route options. This shows up not just in JTW stats but also stats on where people move house from/to – I’m sure Chris could do some visualisations. I think the effect is similar with the Maribyrnong, although perhaps a little less so.
LikeLike
Actually, there is also Burke Road and Banksia Street, but still not that many. I’ve done some visualisations on internal migration that I hope to publish soon.
LikeLike
hello and congratulations for the excellent analysis and super maps, as usual.
i would need to recreate the od maps for an area south of sidney, near port kembla, but i don’t understand the steps to get the data with source / destination pairs. I downloaded the various data but nothing, then I found the data for SA2 but it allows me to map attracted and generated by zone, but not the OD pairs.
could anyone write a step by step tutorial here? it would be very kind and I am sure appreciated by many who have the same problem as me.
Thanks in advance.
LikeLike
Hi Dante, short answer is that you can get OD pairs from the ABS’s tablebuilder pro dataset “counting persons, 15 years and over”. You can access place of enumeration, place of usual residence, and place of work.
LikeLike